
The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells

 ,
,  ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
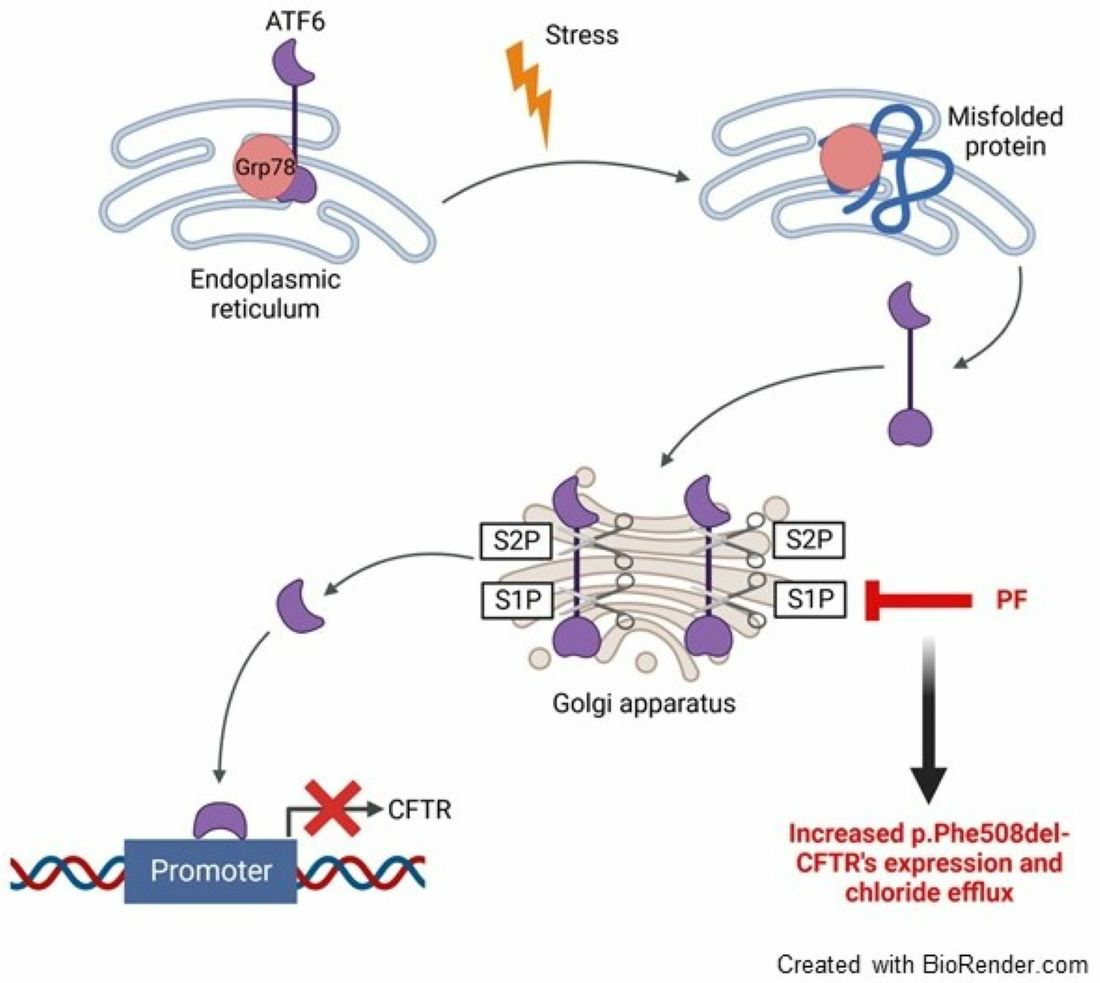
1. Introduction
2. Material and Methods
2.1. Cell Culture and Protein Extraction
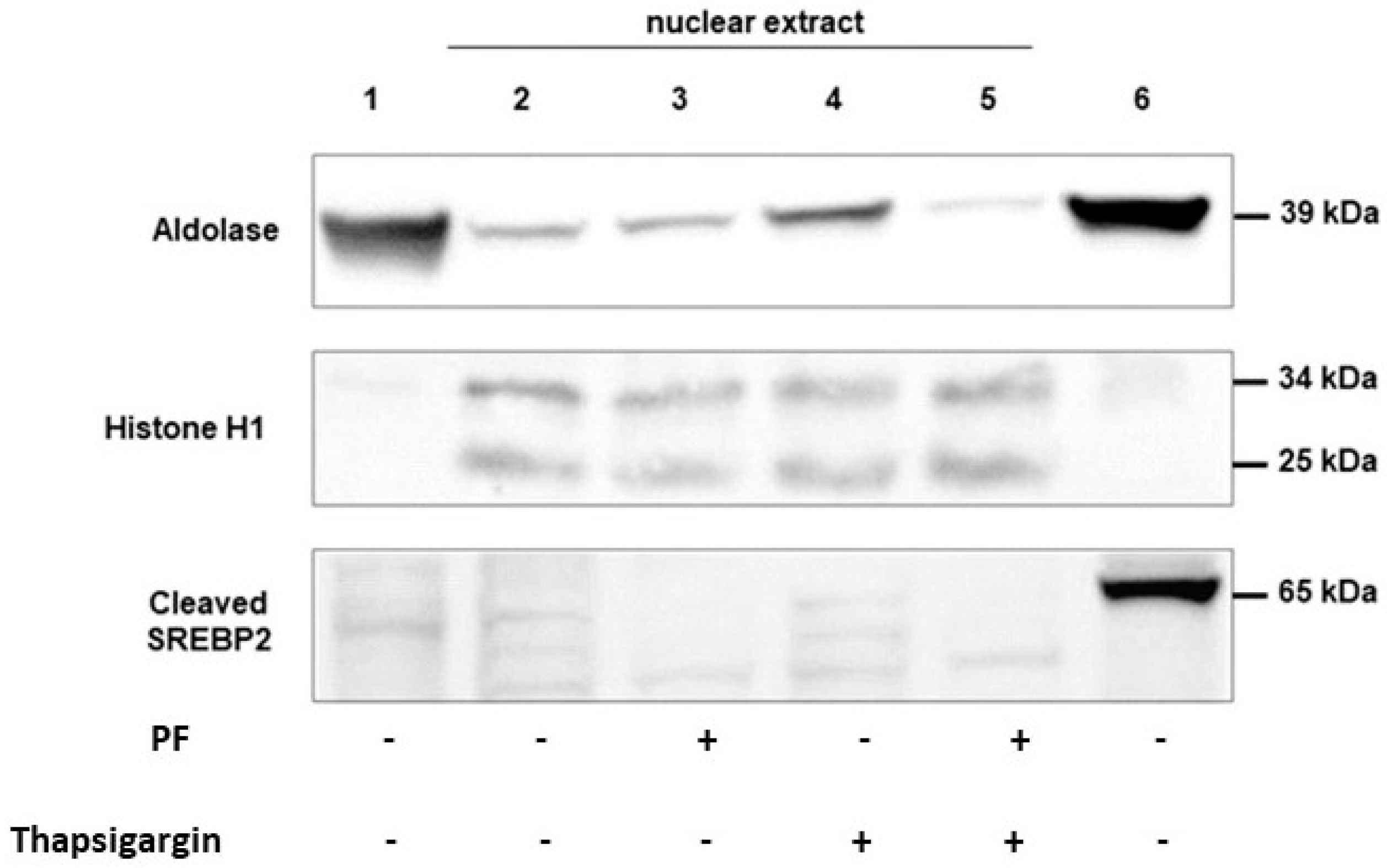
2.2. Nuclear Extract Preparation
2.3. Membrane Extract Preparation
2.4. Immunoprecipitation
2.5. Cell Viability in the Presence of the MBTP1 Inhibitor
2.6. Western Blotting
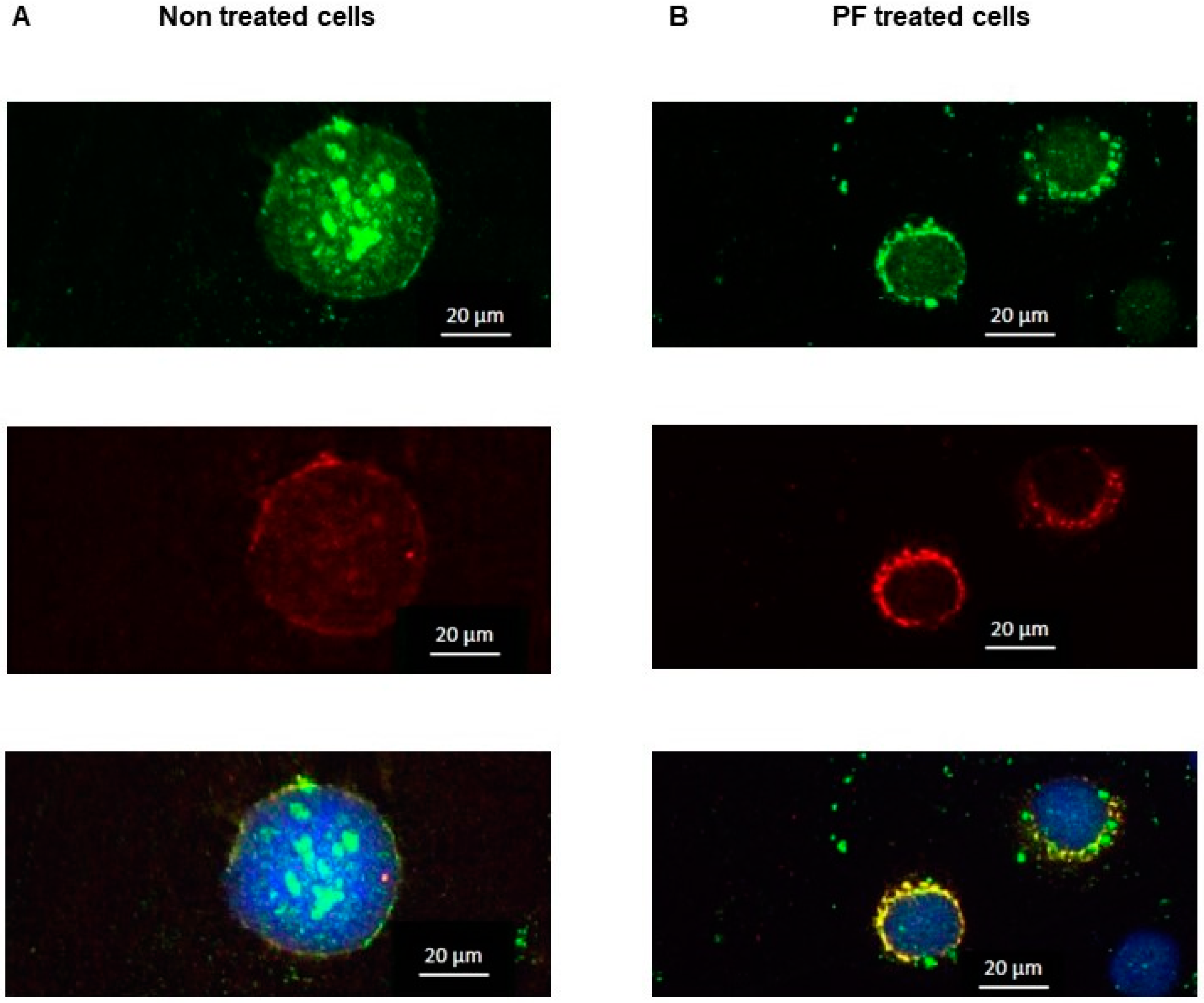
2.7. Immunofluorescence
2.8. RNA Extraction and Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.9. Patch Clamp
2.10. Surface Plasmon Resonance (SPR)
2.11. Gene Expression Analysis Using mRNA Array
2.12. Ussing Chamber Recordings
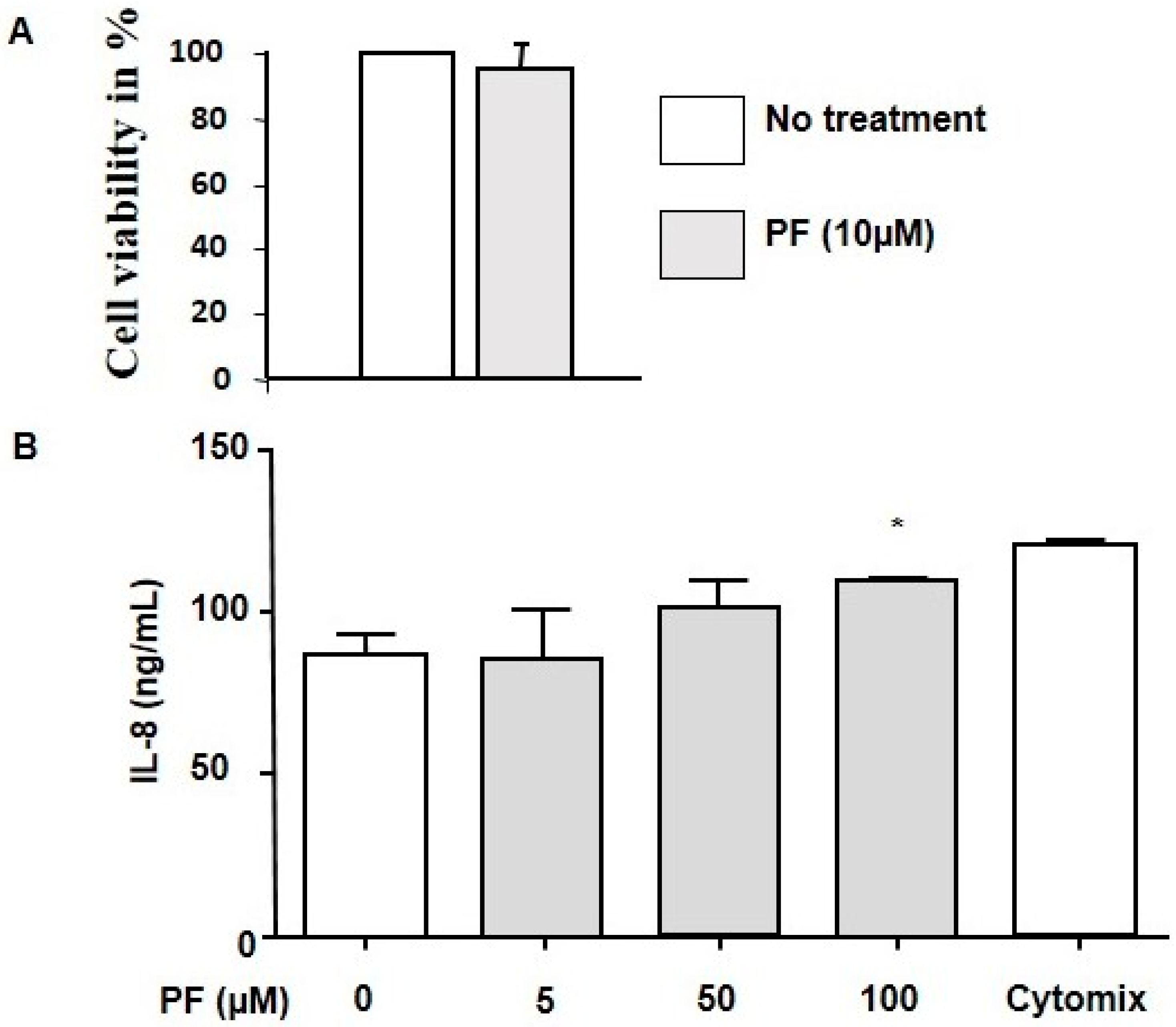
2.13. Interleukin 8 (IL-8) Release
2.14. Statistical Analysis
3. Results
3.1. Cell Viability and Inflammatory Response
3.2. MBTP1 Inhibition Inactivates ATF6 and SREBP2 in CFBE41o-Cells
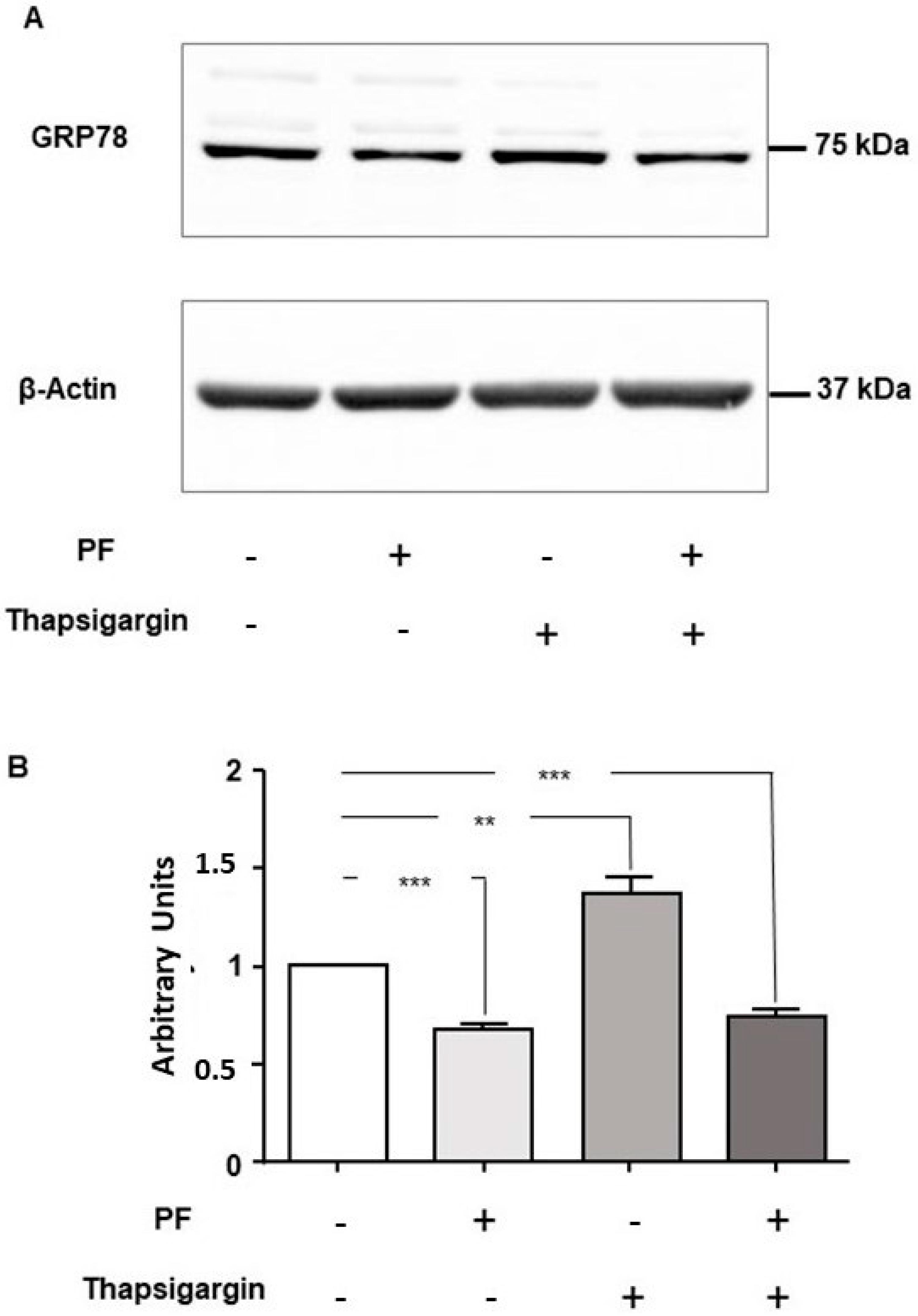
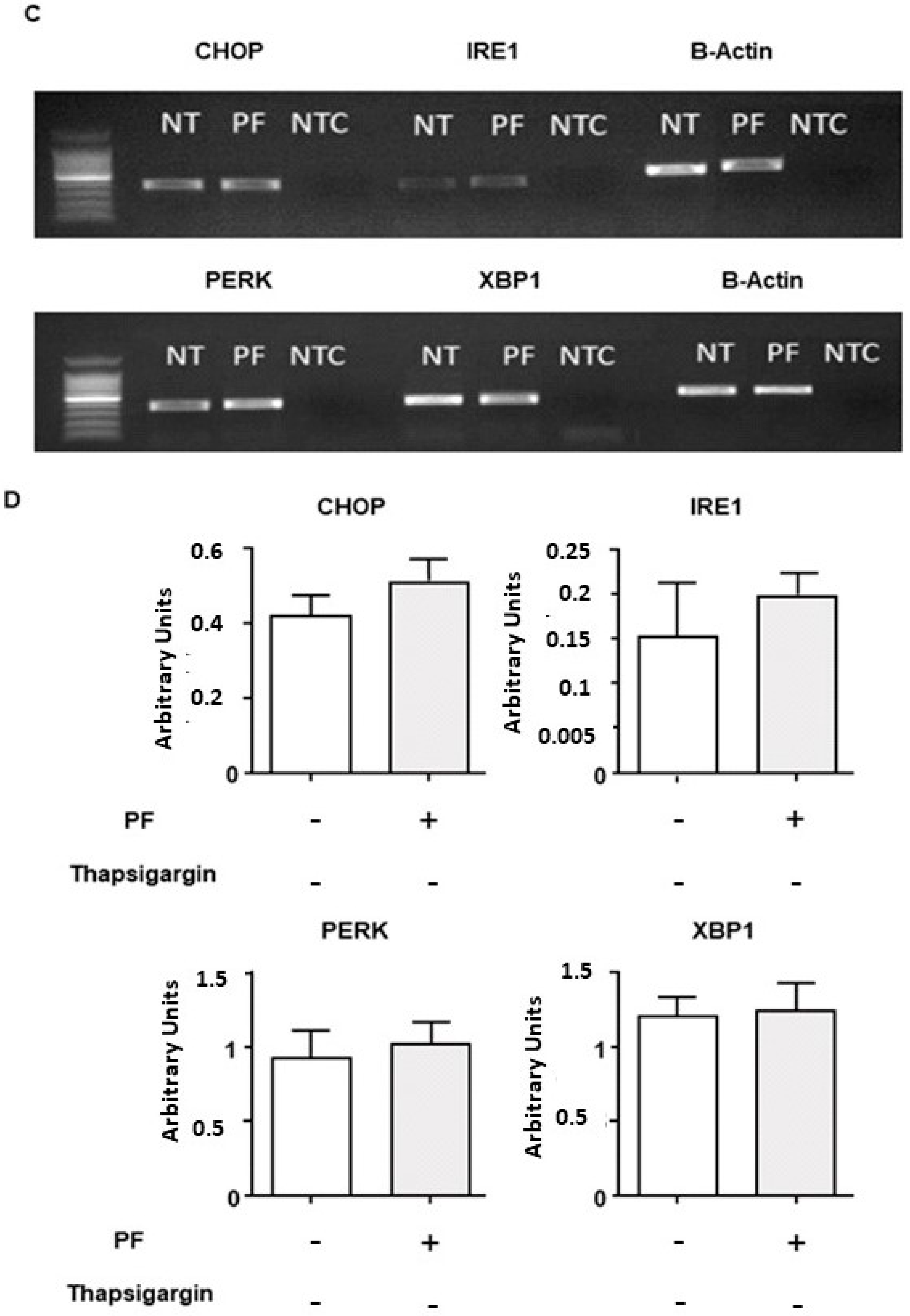
3.3. MBTP1 Inhibition Does Not Trigger the UPR in CFBE41o-Cells
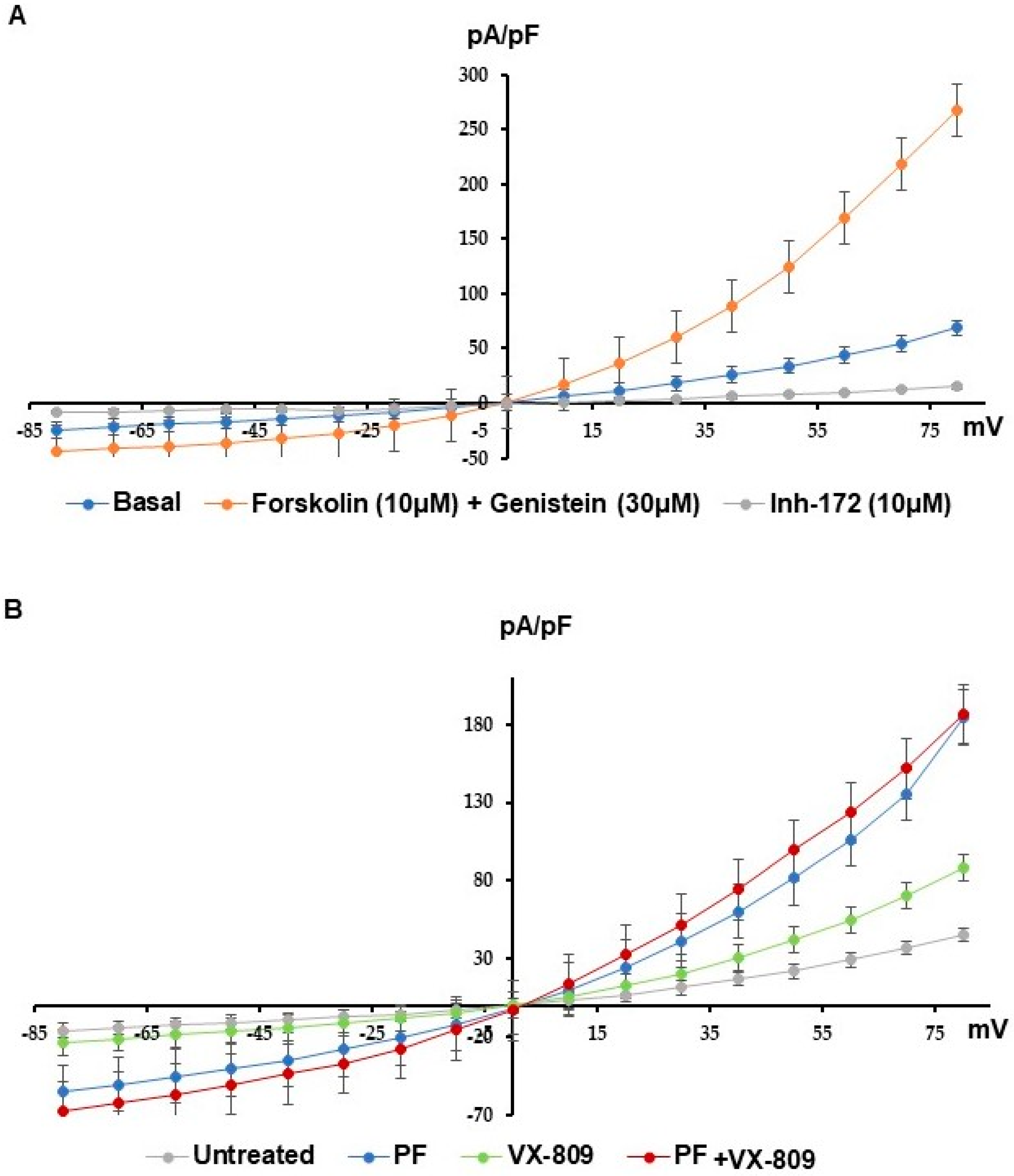
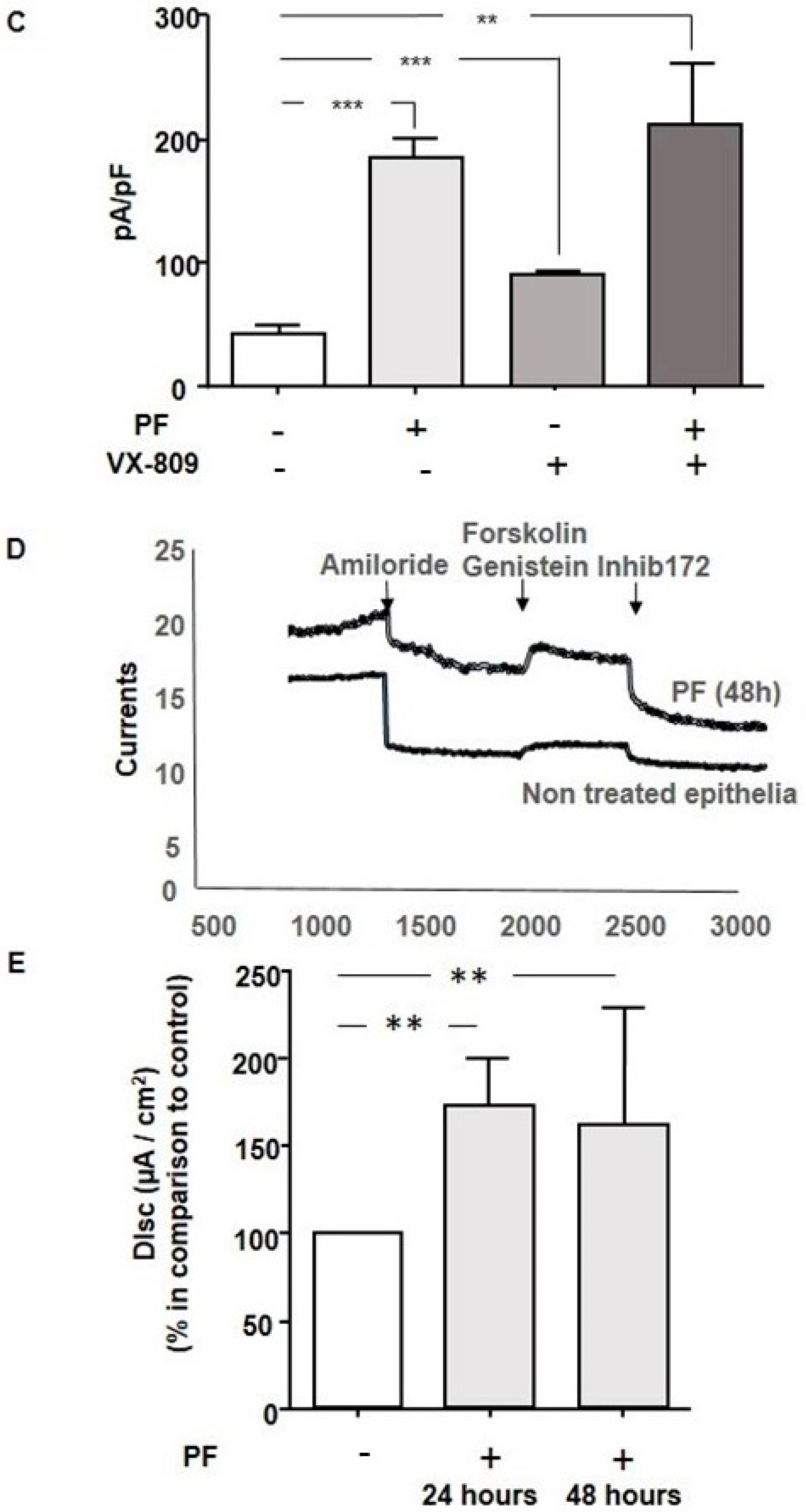
3.4. MBTP1 Inhibition Increases the Cl-Efflux in Cells Expressing p.Phe508del-CFTR
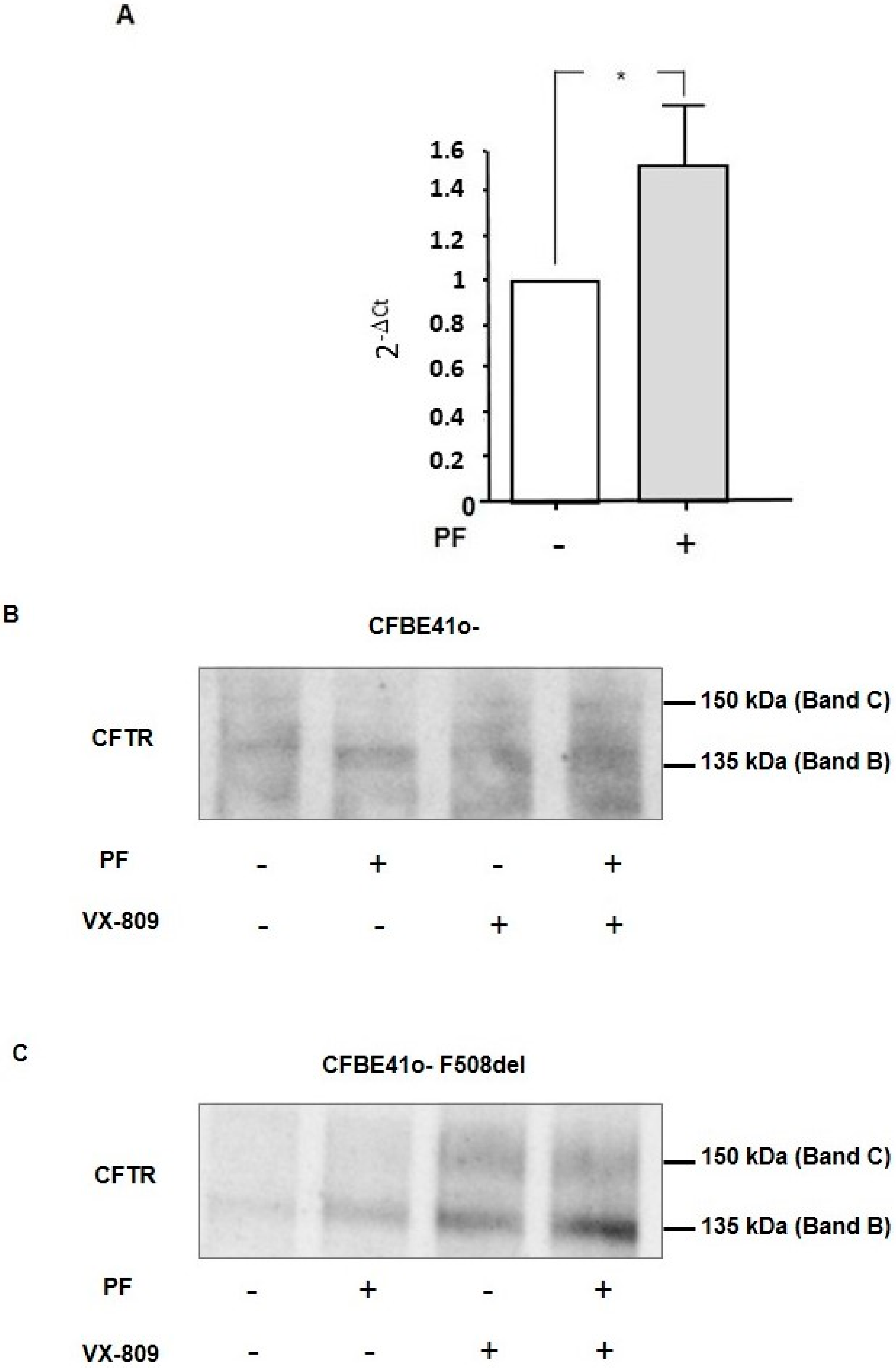
3.5. Inhibition of MBTP1 Increases the Transcription and Protein Expression of p.Phe508del-CFTR
3.6. Inhibition of MBTP1 Increases the Expression of p.Phe508del-CFTR Protein
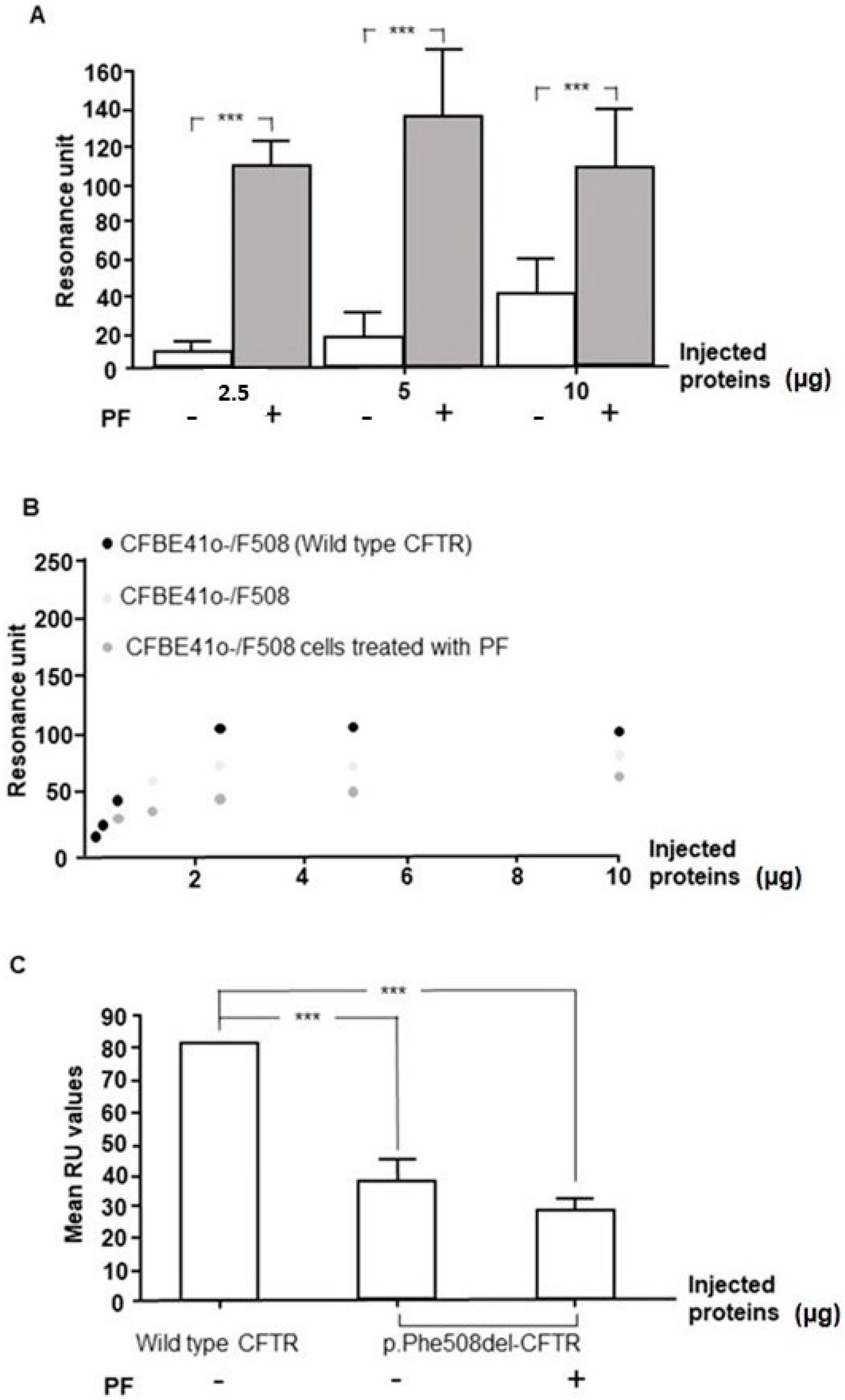
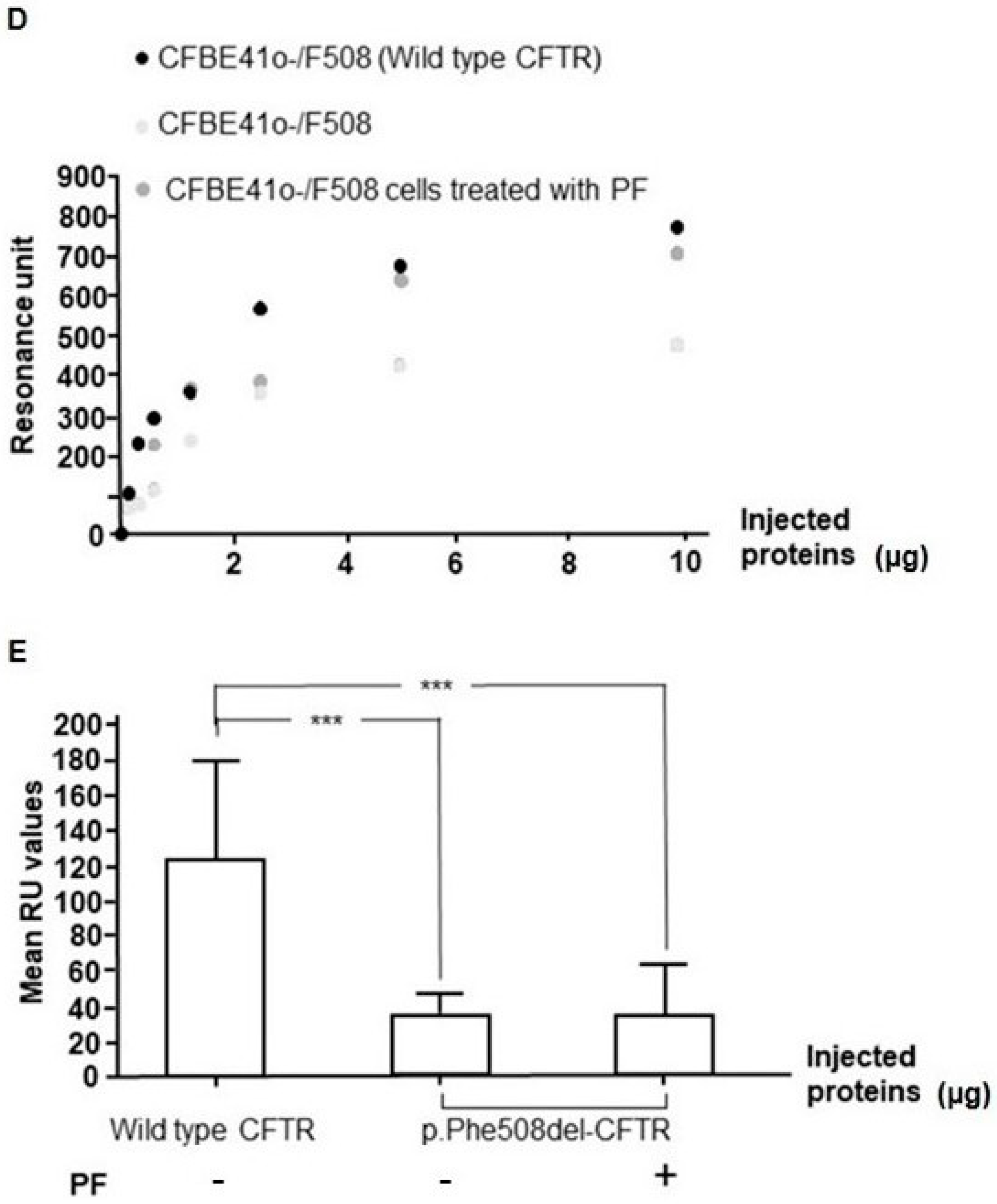
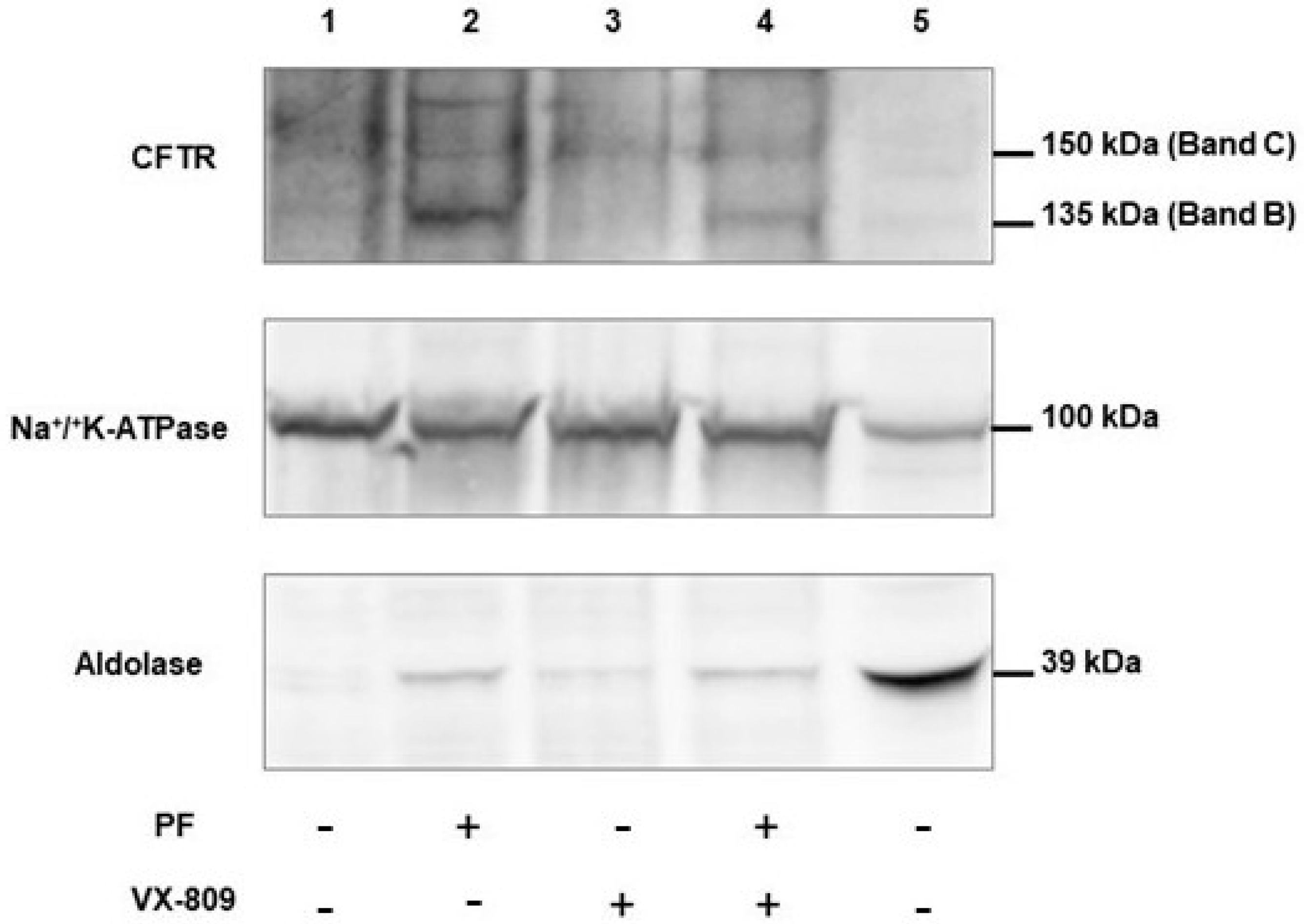
3.7. PF-429242 Increases the Expression of p.Phe508del-CFTR in Membranes
3.8. Comparison of the Gene Expression between Non-Treated and PF-429242-Treated Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kerem, B.S.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rich, D.P.; Anderson, M.P.; Gregory, R.J.; Cheng, S.H.; Paul, S.; Jefferson, D.M.; McCann, J.D.; Klinger, K.W.; Smith, A.E.; Welsh, M.J. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990, 347, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Kothari, V.A.; Park, P.J.; Mulberg, A.E.; Glick, M.C.; Scanlin, T.F. Terminal glycosylation of cystic fibrosis airway epithelial cells. Glycoconj. J. 2000, 17, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991, 253, 202–205. [Google Scholar] [CrossRef]
- Berdiev, B.K.; Qadri, Y.J.; Benos, D.J. Assessment of the CFTR and ENaC association. Mol. Biosyst. 2009, 5, 123–127. [Google Scholar] [CrossRef]
- Du, K.; Sharma, M.; Lukacs, G.L. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat. Struct. Mol. Biol. 2005, 12, 17–25. [Google Scholar] [CrossRef]
- Sharma, M.; Pampinella, F.; Nemes, C.; Benharouga, M.; So, J.; Du, K.; Bache, K.G.; Papsin, B.; Zerangue, N.; Stenmark, H.; et al. Misfolding diverts CFTR from recycling to degradation: Quality control at early endosomes. J. Cell Biol. 2004, 164, 923–933. [Google Scholar] [CrossRef]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar]
- Denning, G.M.; Anderson, M.P.; Amara, J.F.; Marshall, J.; Smith, A.E.; Welsh, M.J. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature 1992, 358, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Grout, M.; Zaidi, T.S.; Olsen, J.C.; Johnson, L.G.; Yankaskas, J.R.; Goldberg, J.B. Role of Mutant CFTR in Hypersusceptibility of Cystic Fibrosis Patients to Lung Infections. Science 1996, 271, 64–67. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Cymberknoh, M.; Kerem, E.; Ferkol, T.; Elizur, A. Airway inflammation in cystic fibrosis: Molecular mechanisms and clinical implications. Thorax 2013, 68, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116 Pt 10, 1861–1862. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef]
- Hai, T.W.; Liu, F.; Coukos, W.J.; Green, M.R. Transcription factor ATF cDNA clones: An extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989, 3, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Baumeister, P.; Roy, B.; Phan, T.; Foti, D.; Luo, S.; Lee, A.S. ATF6 as a transcription activator of the endoplasmic reticulum stress element: Thapsigargin stress-induced changes and synergistic interactions with NF-Y and YY1. Mol. Cell. Biol. 2000, 20, 5096–5106. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Cox, J.S.; Shamu, C.E.; Walter, P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 1993, 73, 1197–1206. [Google Scholar] [CrossRef]
- Zhu, C.; Johansen, F.E.; Prywes, R. Interaction of ATF6 and serum response factor. Mol. Cell. Biol. 1997, 17, 4957–4966. [Google Scholar] [CrossRef]
- Kerbiriou, M.; Le Drévo, M.-A.; Férec, C.; Trouvé, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta 2007, 1772, 1236–1249. [Google Scholar] [CrossRef]
- Rab, A.; Bartoszewski, R.; Jurkuvenaite, A.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am. J. Physiol. Cell Physiol. 2007, 292, C756–C766. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by deltaF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Blohmke, C.J.; Mayer, M.L.; Tang, A.C.; Hirschfeld, A.F.; Fjell, C.D.; Sze, M.A.; Falsafi, R.; Wang, S.; Hsu, K.; Chilvers, M.A.; et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to p38 MAPK-mediated innate immune responses. J. Immunol. 2012, 189, 5467–5475. [Google Scholar] [CrossRef]
- Huguet, F.; Guellec, J.; Kerbiriou, M.; Gandy, M.; Thomas, J.; Férec, C.; Benz, N.; Trouvé, P. Evaluation of aminopyrrolidine amide to improve chloride transport in CFTR-defective cells. Bioorg. Med. Chem. Lett. 2022, 72, 128866. [Google Scholar] [CrossRef]
- Illek, B.; Maurisse, R.; Wahler, L.; Kunzelmann, K.; Fischer, H.; Gruenert, D.C. Cl transport in complemented CF bronchial epithelial cells correlates with CFTR mRNA expression levels. Cell Physiol. Biochem. 2008, 22, 57–68. [Google Scholar] [CrossRef]
- Bruscia, E.; Sangiuolo, F.; Sinibaldi, P.; Goncz, K.K.; Novelli, G.; Gruenert, D.C. Isolation of CF cell lines corrected at DeltaF508-CFTR locus by SFHR-mediated targeting. Gene Ther. 2002, 9, 683–685. [Google Scholar] [CrossRef]
- Hay, B.A.; Abrams, B.; Zumbrunn, A.Y.; Valentine, J.J.; Warren, L.C.; Petras, S.F.; Shelly, L.D.; Xia, A.; Varghese, A.H.; Hawkins, J.L.; et al. Aminopyrrolidineamide inhibitors of site-1 protease. Bioorg. Med. Chem. Lett. 2007, 17, 4411–4414. [Google Scholar] [CrossRef]
- Farre, C.; Stoelzle, S.; Haarmann, C.; George, M.; Brüggemann, A.; Fertig, N. Automated ion channel screening: Patch clamping made easy. Expert Opin. Ther. Targets 2007, 11, 557–565. [Google Scholar] [CrossRef]
- Huguet, F.; Calvez, M.L.; Benz, N.; Le Hir, S.; Mignen, O.; Buscaglia, P.; Horgen, F.D.; Férec, C.; Kerbiriou, M.; Trouvé, P. Function and regulation of TRPM7, as well as intracellular magnesium content, are altered in cells expressing ΔF508-CFTR and G551D-CFTR. Cell Mol. Life Sci. 2016, 73, 3351–3373. [Google Scholar] [CrossRef]
- Trouvé, P.; Calvez, M.-L.; Moisan, S.; Hir, S.L.; Huguet, F.; Benz, N.; Kerbiriou, M.; Férec, C. Rapid detection of the mature form of cystic fibrosis transmembrane regulator by surface plasmon resonance. Anal. Methods 2014, 7, 226–236. [Google Scholar] [CrossRef]
- Inoue, R.; Tsuno, T.; Togashi, Y.; Okuyama, T.; Sato, A.; Nishiyama, K.; Kyohara, M.; Li, J.; Fukushima, S.; Kin, T.; et al. Uncoupling protein 2 and aldolase B impact insulin release by modulating mitochondrial function and Ca2+ release from the ER. iScience 2022, 25, 104603. [Google Scholar] [CrossRef]
- Trouvé, P.; Férec, C.; Génin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Rab, A.; Twitty, G.; Stevenson, L.; Fortenberry, J.; Piotrowski, A.; Dumanski, J.P.; Bebok, Z. The mechanism of cystic fibrosis transmembrane conductance regulator transcriptional repression during the unfolded protein response. J. Biol. Chem. 2008, 283, 12154–12165. [Google Scholar] [CrossRef]
- Caruana, B.T.; Skoric, A.; Brown, A.J.; Lutze-Mann, L.H. Site-1 protease, a novel metabolic target for glioblastoma. Biochem. Biophys. Res. Commun. 2017, 490, 760–766. [Google Scholar] [CrossRef]
- Lin, J.-C.; Liu, T.-P.; Chen, Y.-B.; Yang, P.-M. PF-429242 exhibits anticancer activity in hepatocellular carcinoma cells via FOXO1-dependent autophagic cell death and IGFBP1-dependent anti-survival signaling. Am. J. Cancer Res. 2023, 13, 4125–4144. [Google Scholar]
- Hawkins, J.L.; Robbins, M.D.; Warren, L.C.; Xia, D.; Petras, S.F.; Valentine, J.J.; Varghese, A.H.; Wang, I.-K.; Subashi, T.A.; Shelly, L.D.; et al. Pharmacologic inhibition of site 1 protease activity inhibits sterol regulatory element-binding protein processing and reduces lipogenic enzyme gene expression and lipid synthesis in cultured cells and experimental animals. J. Pharmacol. Exp. Ther. 2008, 326, 801–808. [Google Scholar] [CrossRef]
- Seidah, N.G.; Prat, A. The proprotein convertases are potential targets in the treatment of dyslipidemia. J. Mol. Med. 2007, 85, 685–696. [Google Scholar] [CrossRef]
- Blanchet, M.; Sureau, C.; Guévin, C.; Seidah, N.G.; Labonté, P. SKI-1/S1P inhibitor PF-429242 impairs the onset of HCV infection. Antivir. Res. 2015, 115, 94–104. [Google Scholar] [CrossRef]
- Uchida, L.; Urata, S.; Ulanday, G.E.L.; Takamatsu, Y.; Yasuda, J.; Morita, K.; Hayasaka, D. Suppressive Effects of the Site 1 Protease (S1P) Inhibitor, PF-429242, on Dengue Virus Propagation. Viruses 2016, 8, 46. [Google Scholar] [CrossRef]
- Feng, Y.; Peng, K.; Luo, R.; Wang, F.; Yang, T. Site-1 Protease-Derived Soluble (Pro)Renin Receptor Contributes to Angiotensin II–Induced Hypertension in Mice. Hypertension 2021, 77, 405–416. [Google Scholar] [CrossRef]
- Yuan, Y.; Zhou, Q.; Wang, C.; Zhou, W.; Gu, W.; Zheng, B. Clinical and molecular characterization of a patient with MBTPS1 related spondyloepiphyseal dysplasia: Evidence of pathogenicity for a synonymous variant. Front. Pediatr. 2022, 10, 1056141. [Google Scholar] [CrossRef]
- Kondo, Y.; Fu, J.; Wang, H.; Hoover, C.; McDaniel, J.M.; Steet, R.; Patra, D.; Song, J.; Pollard, L.; Cathey, S.; et al. Site-1 protease deficiency causes human skeletal dysplasia due to defective inter-organelle protein trafficking. JCI Insight 2018, 3, e121596. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Glimcher, L.H. XBP1: The last two decades. Ann. Rheum. Dis. 2010, 69 (Suppl. 1), i67–i71. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef]
- Majumder, M.; Huang, C.; Snider, M.D.; Komar, A.A.; Tanaka, J.; Kaufman, R.J.; Krokowski, D.; Hatzoglou, M. A Novel Feedback Loop Regulates the Response to Endoplasmic Reticulum Stress via the Cooperation of Cytoplasmic Splicing and mRNA Translation. Mol. Cell. Biol. 2012, 32, 992–1003. [Google Scholar] [CrossRef]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef]
- Kim Chiaw, P.; Hantouche, C.; Wong, M.J.H.; Matthes, E.; Robert, R.; Hanrahan, J.W.; Shrier, A.; Young, J.C. Hsp70 and DNAJA2 limit CFTR levels through degradation. PLoS ONE 2019, 14, e0220984. [Google Scholar] [CrossRef]
- Seo, J.H.; Park, J.-H.; Lee, E.J.; Vo, T.T.L.; Choi, H.; Kim, J.Y.; Jang, J.K.; Wee, H.-J.; Lee, H.S.; Jang, S.H.; et al. ARD1-mediated Hsp70 acetylation balances stress-induced protein refolding and degradation. Nat. Commun. 2016, 7, 12882. [Google Scholar] [CrossRef]
- Pittman, N.; Shue, G.; LeLeiko, N.S.; Walsh, M.J. Transcription of cystic fibrosis transmembrane conductance regulator requires a CCAAT-like element for both basal and cAMP-mediated regulation. J. Biol. Chem. 1995, 270, 28848–28857. [Google Scholar] [CrossRef]
- NandyMazumdar, M.; Yin, S.; Paranjapye, A.; Kerschner, J.L.; Swahn, H.; Ge, A.; Leir, S.-H.; Harris, A. Looping of upstream cis-regulatory elements is required for CFTR expression in human airway epithelial cells. Nucleic Acids Res. 2020, 48, 3513–3524. [Google Scholar] [CrossRef]
- Collawn, J.F.; Fu, L.; Bebok, Z. Targets for cystic fibrosis therapy: Proteomic analysis and correction of mutant cystic fibrosis transmembrane conductance regulator. Expert Rev. Proteom. 2010, 7, 495–506. [Google Scholar] [CrossRef]
- Baudouin-Legros, M.; Hinzpeter, A.; Jaulmes, A.; Brouillard, F.; Costes, B.; Fanen, P.; Edelman, A. Cell-specific posttranscriptional regulation of CFTR gene expression via influence of MAPK cascades on 3’UTR part of transcripts. Am. J. Physiol. Cell Physiol. 2005, 289, C1240–C1250. [Google Scholar] [CrossRef]
- Xu, H.-D.; Qin, Z.-H. Beclin 1, Bcl-2 and Autophagy. In Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology; Qin, Z.-H., Ed.; Springer: Singapore, 2019; pp. 109–126. [Google Scholar] [CrossRef]
- Bhardwaj, M.; Leli, N.M.; Koumenis, C.; Amaravadi, R.K. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin. Cancer Biol. 2020, 66, 116–128. [Google Scholar] [CrossRef]
- Park, H.; Shin, D.H.; Sim, J.-R.; Aum, S.; Lee, M.G. IRE1α kinase-mediated unconventional protein secretion rescues misfolded CFTR and pendrin. Sci. Adv. 2020, 6, eaax9914. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef]
- Lee, Y.-Y.; Cevallos, R.C.; Jan, E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J. Biol. Chem. 2009, 284, 6661–6673. [Google Scholar] [CrossRef]
- Brush, M.H.; Shenolikar, S. Control of cellular GADD34 levels by the 26S proteasome. Mol. Cell. Biol. 2008, 28, 6989–7000. [Google Scholar] [CrossRef]
- Bridges, J.P.; Schehr, A.; Wang, Y.; Huo, L.; Besnard, V.; Ikegami, M.; Whitsett, J.A.; Xu, Y. Epithelial SCAP/INSIG/SREBP signaling regulates multiple biological processes during perinatal lung maturation. PLoS ONE 2014, 9, e91376. [Google Scholar] [CrossRef]
- McPherson, R.; Gauthier, A. Molecular regulation of SREBP function: The Insig-SCAP connection and isoform-specific modulation of lipid synthesis. Biochem. Cell Biol. 2004, 82, 201–211. [Google Scholar] [CrossRef]
- Ernst, W.L.; Shome, K.; Wu, C.C.; Gong, X.; Frizzell, R.A.; Aridor, M. VAMP-associated Proteins (VAP) as Receptors That Couple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Proteostasis with Lipid Homeostasis. J. Biol. Chem. 2016, 291, 5206–5220. [Google Scholar] [CrossRef]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Cottrill, K.A.; Farinha, C.M.; McCarty, N.A. The bidirectional relationship between CFTR and lipids. Commun. Biol. 2020, 3, 179. [Google Scholar] [CrossRef]
- Baiceanu, A.; Mesdom, P.; Lagouge, M.; Foufelle, F. Endoplasmic reticulum proteostasis in hepatic steatosis. Nat. Rev. Endocrinol. 2016, 12, 710–722. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VX-445–Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Aalbers, B.L.; de Winter-de Groot, K.M.; Arets, H.G.M.; Hofland, R.W.; de Kiviet, A.C.; van Oirschot-van de Ven, M.M.M.; Kruijswijk, M.A.; Schotman, S.; Michel, S.; van der Ent, C.K.; et al. Clinical effect of lumacaftor/ivacaftor in F508del homozygous CF patients with FEV1 ≥ 90% predicted at baseline. J. Cyst. Fibros. 2020, 19, 654–658. [Google Scholar] [CrossRef]
- Lo Cicero, S.; Castelli, G.; Blaconà, G.; Bruno, S.M.; Sette, G.; Pigliucci, R.; Villella, V.R.; Esposito, S.; Zollo, I.; Spadaro, F.; et al. L1077P CFTR pathogenic variant function rescue by Elexacaftor–Tezacaftor–Ivacaftor in cystic fibrosis patient-derived air–liquid interface (ALI) cultures and organoids: In vitro guided personalized therapy of non-F508del patients. Respir. Res. 2023, 24, 217. [Google Scholar] [CrossRef]
- Harutyunyan, M.; Huang, Y.; Mun, K.-S.; Yang, F.; Arora, K.; Naren, A.P. Personalized medicine in CF: From modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L529–L543. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santinelli, R.; Benz, N.; Guellec, J.; Quinquis, F.; Kocas, E.; Thomas, J.; Montier, T.; Ka, C.; Luczka-Majérus, E.; Sage, E.; et al. The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells. Cells 2024, 13, 185. https://doi.org/10.3390/cells13020185
Santinelli R, Benz N, Guellec J, Quinquis F, Kocas E, Thomas J, Montier T, Ka C, Luczka-Majérus E, Sage E, et al. The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells. Cells. 2024; 13(2):185. https://doi.org/10.3390/cells13020185
Chicago/Turabian StyleSantinelli, Raphaël, Nathalie Benz, Julie Guellec, Fabien Quinquis, Ervin Kocas, Johan Thomas, Tristan Montier, Chandran Ka, Emilie Luczka-Majérus, Edouard Sage, and et al. 2024. "The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells" Cells 13, no. 2: 185. https://doi.org/10.3390/cells13020185
APA StyleSantinelli, R., Benz, N., Guellec, J., Quinquis, F., Kocas, E., Thomas, J., Montier, T., Ka, C., Luczka-Majérus, E., Sage, E., Férec, C., Coraux, C., & Trouvé, P. (2024). The Inhibition of the Membrane-Bound Transcription Factor Site-1 Protease (MBTP1) Alleviates the p.Phe508del-Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Defects in Cystic Fibrosis Cells. Cells, 13(2), 185. https://doi.org/10.3390/cells13020185