
AIBP: A New Safeguard against Glaucomatous Neuroinflammation

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
Abstract
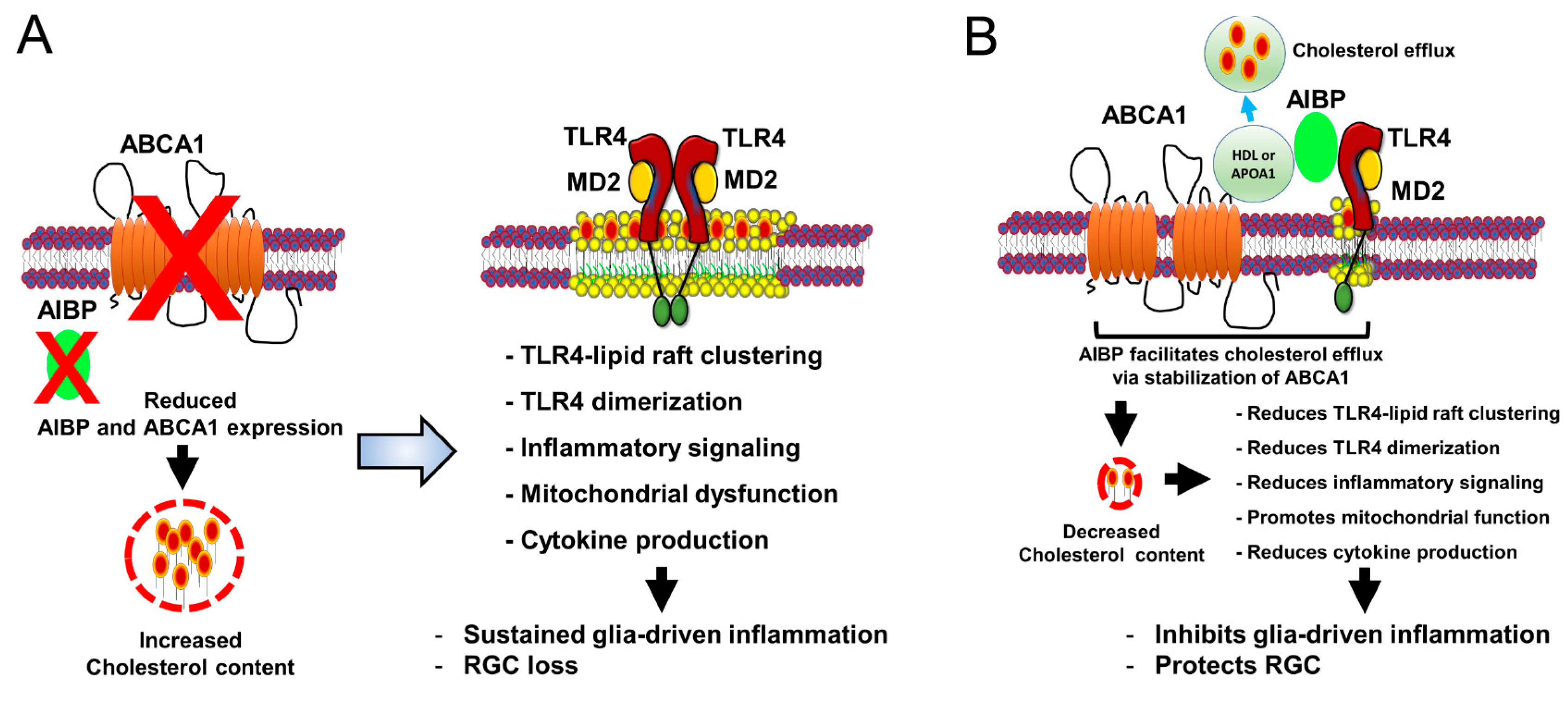
:1. Introduction
2. Potential Role of AIBP in Lipid Rafts and Cholesterol Metabolism in Glaucomatous Retina
3. Role of AIBP in Regulation of Glial-Driven Retinal Neuroinflammation and RGC Degeneration
4. Role of AIBP in Mitochondrial Dynamics and Function in Glaucomatous Neuroinflammation and Neurodegeneration
5. AIBP-Mediated Neuroprotection in Glaucomatous Neuroinflammation and Neurodegeneration
6. Potential Role of AIBP in the NAD(P)HX Repair System of the Retina
7. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| AAV | adeno-associated virus |
| ABCA1 | ATP-binding cassette transporter A1 |
| AIBP | apolipoprotein A-I-binding protein |
| AMPK | AMP-activated protein kinase |
| APOA-I | apolipoprotein A-I |
| ATP | adenosine triphosphate |
| BBB | blood–brain barrier |
| DAMP | damage-associated molecular pattern |
| DRP1 | dynamin-related protein 1 |
| HDL | high-density lipoprotein |
| IOP | intraocular pressure |
| LDL | low-density lipoprotein |
| LPS | lipopolysaccharide |
| MFN | mitofusin |
| MMP | mitochondrial membrane potential |
| mtDAMP | mitochondrial damage-associated molecular pattern |
| NAD | nicotinamide adenine dinucleotide |
| NADP | nicotinamide adenine dinucleotide phosphate |
| NAM | nicotinamide |
| NAXE | NAD(P)H-X epimerase |
| NMN | nicotinamide mononucleotide |
| ONC | optic nerve crush |
| OPA1 | optic atrophy type 1 |
| OXPHOS | oxidative phosphorylation |
| PAMP | pathogen-associated molecular pattern |
| PARK2 | Parkin |
| POAG | primary open-angle glaucoma |
| PRR | pathogen recognition receptor |
| RGC | retinal ganglion cell |
| ROS | reactive oxygen species |
| RPE | retinal pigment epithelium |
| SNP | single nucleotide polymorphism |
| TLR4 | toll-like receptor 4 |
References
- Weinreb, R.N.; Khaw, P.T. Primary open-angle glaucoma. Lancet 2004, 363, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The pathophysiology and treatment of glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Aung, T.; Bourne, R.R.; Bron, A.M.; Ritch, R.; Panda-Jonas, S. Glaucoma. Lancet 2017, 390, 2183–2193. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Perkins, G.A.; Kim, K.Y.; Bastola, T.; Choi, W.Y.; Choi, S.H. Glaucomatous optic neuropathy: Mitochondrial dynamics, dysfunction and protection in retinal ganglion cells. Prog. Retin. Eye Res. 2022, 95, 101136. [Google Scholar] [CrossRef] [PubMed]
- Tezel, G. Multifactorial Pathogenic Processes of Retinal Ganglion Cell Degeneration in Glaucoma towards Multi-Target Strategies for Broader Treatment Effects. Cells 2021, 10, 1372. [Google Scholar] [CrossRef] [PubMed]
- Nemesure, B.; Honkanen, R.; Hennis, A.; Wu, S.Y.; Leske, M.C.; Barbados Eye Studies, G. Incident open-angle glaucoma and intraocular pressure. Ophthalmology 2007, 114, 1810–1815. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Leung, C.K.; Crowston, J.G.; Medeiros, F.A.; Friedman, D.S.; Wiggs, J.L.; Martin, K.R. Primary open-angle glaucoma. Nat. Rev. Dis. Primers 2016, 2, 16067. [Google Scholar] [CrossRef]
- Hirt, J.; Porter, K.; Dixon, A.; McKinnon, S.; Liton, P.B. Contribution of autophagy to ocular hypertension and neurodegeneration in the DBA/2J spontaneous glaucoma mouse model. Cell Death Discov. 2018, 4, 14. [Google Scholar] [CrossRef]
- Qu, J.; Wang, D.; Grosskreutz, C.L. Mechanisms of retinal ganglion cell injury and defense in glaucoma. Exp. Eye Res. 2010, 91, 48–53. [Google Scholar] [CrossRef]
- Nickells, R.W. From ocular hypertension to ganglion cell death: A theoretical sequence of events leading to glaucoma. Can. J. Ophthalmol. 2007, 42, 278–287. [Google Scholar]
- Choi, S.H.; Kim, K.Y.; Perkins, G.A.; Phan, S.; Edwards, G.; Xia, Y.; Kim, J.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Ellisman, M.H.; et al. AIBP protects retinal ganglion cells against neuroinflammation and mitochondrial dysfunction in glaucomatous neurodegeneration. Redox Biol. 2020, 37, 101703. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Ha, Y.; Choi, S.; Kim, K.Y.; Bastola, T.; Kim, J.; Weinreb, R.N.; Zhang, W.; Miller, Y.I.; Choi, S.H. Restoring AIBP deficiency in the retina provides neuroprotection in glaucoma. bioRxiv 2023, preprint. [Google Scholar] [CrossRef]
- Ritter, M.; Buechler, C.; Boettcher, A.; Barlage, S.; Schmitz-Madry, A.; Orso, E.; Bared, S.M.; Schmiedeknecht, G.; Baehr, C.H.; Fricker, G.; et al. Cloning and characterization of a novel apolipoprotein A-I binding protein, AI-BP, secreted by cells of the kidney proximal tubules in response to HDL or ApoA-I. Genomics 2002, 79, 693–702. [Google Scholar] [CrossRef]
- Fang, L.; Choi, S.H.; Baek, J.S.; Liu, C.; Almazan, F.; Ulrich, F.; Wiesner, P.; Taleb, A.; Deer, E.; Pattison, J.; et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature 2013, 498, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.A.; Choi, S.H.; Agatisa-Boyle, C.; Zhu, L.; Kim, J.; Pattison, J.; Sears, D.D.; Gordts, P.; Fang, L.; Miller, Y.I. AIBP protects against metabolic abnormalities and atherosclerosis. J. Lipid Res. 2018, 59, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Woller, S.A.; Choi, S.H.; An, E.J.; Low, H.; Schneider, D.A.; Ramachandran, R.; Kim, J.; Bae, Y.S.; Sviridov, D.; Corr, M.; et al. Inhibition of Neuroinflammation by AIBP: Spinal Effects upon Facilitated Pain States. Cell Rep. 2018, 23, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Wallace, A.M.; Schneider, D.A.; Burg, E.; Kim, J.; Alekseeva, E.; Ubags, N.D.; Cool, C.D.; Fang, L.; Suratt, B.T.; et al. AIBP augments cholesterol efflux from alveolar macrophages to surfactant and reduces acute lung inflammation. JCI Insight 2018, 3, e120519. [Google Scholar] [CrossRef]
- Navia-Pelaez, J.M.; Choi, S.-H.; dos Santos Aggum Capettini, L.; Xia, Y.; Gonen, A.; Agatisa-Boyle, C.; Delay, L.; Gonçalves dos Santos, G.; Catroli, G.F.; Kim, J.; et al. Normalization of cholesterol metabolism in spinal microglia alleviates neuropathic pain. J. Exp. Med. 2021, 218, e20202059. [Google Scholar] [CrossRef]
- Choi, S.H.; Agatisa-Boyle, C.; Gonen, A.; Kim, A.; Kim, J.; Alekseeva, E.; Tsimikas, S.; Miller, Y.I. Intracellular AIBP (Apolipoprotein A-I Binding Protein) Regulates Oxidized LDL (Low-Density Lipoprotein)-Induced Mitophagy in Macrophages. Arterioscler. Thromb. Vasc. Biol. 2021, 41, e82–e96. [Google Scholar] [CrossRef]
- Zhang, M.; Zhao, G.J.; Yin, K.; Xia, X.D.; Gong, D.; Zhao, Z.W.; Chen, L.Y.; Zheng, X.L.; Tang, X.E.; Tang, C.K. Apolipoprotein A-1 Binding Protein Inhibits Inflammatory Signaling Pathways by Binding to Apolipoprotein A-1 in THP-1 Macrophages. Circ. J. 2018, 82, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Navia-Pelaez, J.M.; Borges Paes Lemes, J.; Gonzalez, L.; Delay, L.; Capettini, L.D.S.A.; Lu, J.W.; Goncalves Dos Santos, G.; Gregus, A.M.; Dougherty, P.M.; Yaksh, T.L.; et al. AIBP regulates TRPV1 activation in chemotherapy-induced peripheral neuropathy by controlling lipid raft dynamics and proximity to TLR4 in dorsal root ganglion neurons. Pain 2023, 164, e274–e285. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Feringa, F.M.; van der Kant, R. Cholesterol and Alzheimer’s Disease; From Risk Genes to Pathological Effects. Front. Aging Neurosci. 2021, 13, 690372. [Google Scholar] [CrossRef] [PubMed]
- Gabrielle, P.H. Lipid metabolism and retinal diseases. Acta Ophthalmol. 2022, 100 (Suppl. S269), 3–43. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Navia-Pelaez, J.M.; Corr, M.; Yaksh, T.L. Lipid rafts in glial cells: Role in neuroinflammation and pain processing. J. Lipid Res. 2020, 61, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Owen, J.S.; Wilson, M.D.; Li, H.; Griffiths, G.L.; Thomas, M.J.; Hiltbold, E.M.; Fessler, M.B.; Parks, J.S. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J. Lipid Res. 2010, 51, 3196–3206. [Google Scholar] [CrossRef]
- Liu, B.; He, Z.; Wang, J.; Xin, Z.; Wang, J.; Li, F.; Fu, Y. Taraxasterol Inhibits LPS-Induced Inflammatory Response in BV2 Microglia Cells by Activating LXRalpha. Front. Pharmacol. 2018, 9, 278. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, Y.; Vithana, E.N.; Jia, L.; Zuo, X.; Wong, T.Y.; Chen, L.J.; Zhu, X.; Tam, P.O.; Gong, B.; et al. Common variants near ABCA1 and in PMM2 are associated with primary open-angle glaucoma. Nat. Genet. 2014, 46, 1115–1119. [Google Scholar] [CrossRef]
- Bailey, J.N.; Loomis, S.J.; Kang, J.H.; Allingham, R.R.; Gharahkhani, P.; Khor, C.C.; Burdon, K.P.; Aschard, H.; Chasman, D.I.; Igo, R.P., Jr.; et al. Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat. Genet. 2016, 48, 189–194. [Google Scholar] [CrossRef]
- van Koolwijk, L.M.; Ramdas, W.D.; Ikram, M.K.; Jansonius, N.M.; Pasutto, F.; Hysi, P.G.; Macgregor, S.; Janssen, S.F.; Hewitt, A.W.; Viswanathan, A.C.; et al. Common genetic determinants of intraocular pressure and primary open-angle glaucoma. PLoS Genet. 2012, 8, e1002611. [Google Scholar] [CrossRef]
- Wiggs, J.L.; Pasquale, L.R. Genetics of glaucoma. Hum. Mol. Genet. 2017, 26, R21–R27. [Google Scholar] [CrossRef] [PubMed]
- Gharahkhani, P.; Burdon, K.P.; Fogarty, R.; Sharma, S.; Hewitt, A.W.; Martin, S.; Law, M.H.; Cremin, K.; Bailey, J.N.C.; Loomis, S.J.; et al. Common variants near ABCA1, AFAP1 and GMDS confer risk of primary open-angle glaucoma. Nat. Genet. 2014, 46, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- Hysi, P.G.; Cheng, C.Y.; Springelkamp, H.; Macgregor, S.; Bailey, J.N.C.; Wojciechowski, R.; Vitart, V.; Nag, A.; Hewitt, A.W.; Hohn, R.; et al. Genome-wide analysis of multi-ancestry cohorts identifies new loci influencing intraocular pressure and susceptibility to glaucoma. Nat. Genet. 2014, 46, 1126–1130. [Google Scholar] [CrossRef]
- Shiga, Y.; Akiyama, M.; Nishiguchi, K.M.; Sato, K.; Shimozawa, N.; Takahashi, A.; Momozawa, Y.; Hirata, M.; Matsuda, K.; Yamaji, T.; et al. Genome-wide association study identifies seven novel susceptibility loci for primary open-angle glaucoma. Hum. Mol. Genet. 2018, 27, 1486–1496. [Google Scholar] [CrossRef]
- Shinozaki, Y.; Leung, A.; Namekata, K.; Saitoh, S.; Nguyen, H.B.; Takeda, A.; Danjo, Y.; Morizawa, Y.M.; Shigetomi, E.; Sano, F.; et al. Astrocytic dysfunction induced by ABCA1 deficiency causes optic neuropathy. Sci. Adv. 2022, 8, eabq1081. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, S.; Zhou, Z.; Zhao, Y. Ad- and AAV8-mediated ABCA1 gene therapy in a murine model with retinal ischemia/reperfusion injuries. Mol. Ther. Methods Clin. Dev. 2021, 20, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhao, G.J.; Yao, F.; Xia, X.D.; Gong, D.; Zhao, Z.W.; Chen, L.Y.; Zheng, X.L.; Tang, X.E.; Tang, C.K. AIBP reduces atherosclerosis by promoting reverse cholesterol transport and ameliorating inflammation in apoE−/− mice. Atherosclerosis 2018, 273, 122–130. [Google Scholar] [CrossRef]
- Zhu, L.; Parker, M.; Enemchukwu, N.; Shen, M.; Zhang, G.; Yan, Q.; Handa, J.T.; Fang, L.; Fu, Y. Combination of apolipoprotein-A-I/apolipoprotein-A-I binding protein and anti-VEGF treatment overcomes anti-VEGF resistance in choroidal neovascularization in mice. Commun. Biol. 2020, 3, 386. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, Q.; Wang, Y.; Wang, J.; Su, Y.; Wang, F.; Wang, G. AIBP and APOA-I synergistically inhibit intestinal tumor growth and metastasis by promoting cholesterol efflux. J. Transl. Med. 2019, 17, 161. [Google Scholar] [CrossRef]
- Sun, B.; Yu, L.; Xu, C.; Li, Y.M.; Zhao, Y.R.; Cao, M.M.; Yang, L.Y. NAD(P)HX epimerase downregulation promotes tumor progression through ROS/HIF-1alpha signaling in hepatocellular carcinoma. Cancer Sci. 2021, 112, 2753–2769. [Google Scholar] [CrossRef] [PubMed]
- Dietschy, J.M.; Turley, S.D. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 2001, 12, 105–112. [Google Scholar] [CrossRef]
- Nes, W.D. Biosynthesis of cholesterol and other sterols. Chem. Rev. 2011, 111, 6423–6451. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.A.; Weickert, C.S.; Garner, B. Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin. Lipidol. 2010, 51, 555–573. [Google Scholar] [CrossRef] [PubMed]
- Stukas, S.; Robert, J.; Lee, M.; Kulic, I.; Carr, M.; Tourigny, K.; Fan, J.; Namjoshi, D.; Lemke, K.; DeValle, N.; et al. Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J. Am. Heart Assoc. 2014, 3, e001156. [Google Scholar] [CrossRef] [PubMed]
- Wellington, C.L.; Frikke-Schmidt, R. Relation between plasma and brain lipids. Curr. Opin. Lipidol. 2016, 27, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Luo, C.; Zhao, J.; Devarajan, G.; Xu, H. Immune regulation in the aging retina. Prog. Retin. Eye Res. 2019, 69, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.G.; Bailey, K.R.; Kane, J.P.; Schwartz, D.M. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem. Biophys. Res. Commun. 2002, 292, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.G.; Hosseini, K.; Bailey, K.R.; Yang, H.; Lowe, R.J.; Matthes, M.T.; Kane, J.P.; LaVail, M.M.; Schwartz, D.M.; Duncan, J.L. Expression of reverse cholesterol transport proteins ATP-binding cassette A1 (ABCA1) and scavenger receptor BI (SR-BI) in the retina and retinal pigment epithelium. Br. J. Ophthalmol. 2009, 93, 1116–1120. [Google Scholar] [CrossRef]
- Fliesler, S.J.; Bretillon, L. The ins and outs of cholesterol in the vertebrate retina. J. Lipid Res. 2010, 51, 3399–3413. [Google Scholar] [CrossRef]
- Tserentsoodol, N.; Gordiyenko, N.V.; Pascual, I.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Intraretinal lipid transport is dependent on high density lipoprotein-like particles and class B scavenger receptors. Mol. Vis. 2006, 12, 1319–1333. [Google Scholar] [PubMed]
- Tserentsoodol, N.; Sztein, J.; Campos, M.; Gordiyenko, N.V.; Fariss, R.N.; Lee, J.W.; Fliesler, S.J.; Rodriguez, I.R. Uptake of cholesterol by the retina occurs primarily via a low density lipoprotein receptor-mediated process. Mol. Vis. 2006, 12, 1306–1318. [Google Scholar] [PubMed]
- Posch-Pertl, L.; Michelitsch, M.; Wagner, G.; Wildner, B.; Silbernagel, G.; Pregartner, G.; Wedrich, A. Cholesterol and glaucoma: A systematic review and meta-analysis. Acta Ophthalmol. 2022, 100, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Welch, C.; Pagler, T.A.; Ranalletta, M.; Lamkanfi, M.; Han, S.; Ishibashi, M.; Li, R.; Wang, N.; Tall, A.R. Increased inflammatory gene expression in ABC transporter-deficient macrophages: Free cholesterol accumulation, increased signaling via toll-like receptors, and neutrophil infiltration of atherosclerotic lesions. Circulation 2008, 118, 1837–1847. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ishibashi, M.; Seimon, T.; Lee, M.; Sharma, S.M.; Fitzgerald, K.A.; Samokhin, A.O.; Wang, Y.; Sayers, S.; Aikawa, M.; et al. Free cholesterol accumulation in macrophage membranes activates Toll-like receptors and p38 mitogen-activated protein kinase and induces cathepsin K. Circ. Res. 2009, 104, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef]
- Zhu, X.; Lee, J.Y.; Timmins, J.M.; Brown, J.M.; Boudyguina, E.; Mulya, A.; Gebre, A.K.; Willingham, M.C.; Hiltbold, E.M.; Mishra, N.; et al. Increased cellular free cholesterol in macrophage-specific Abca1 knock-out mice enhances pro-inflammatory response of macrophages. J. Biol. Chem. 2008, 283, 22930–22941. [Google Scholar] [CrossRef]
- Murphy, A.J.; Woollard, K.J.; Hoang, A.; Mukhamedova, N.; Stirzaker, R.A.; McCormick, S.P.; Remaley, A.T.; Sviridov, D.; Chin-Dusting, J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2071–2077. [Google Scholar] [CrossRef]
- Chen, R.X.; Jiang, W.J.; Liu, S.C.; Wang, Z.Y.; Wang, Z.B.; Zhou, T.; Chen, Y.A.; Wang, J.F.; Chang, J.; Wang, Y.R.; et al. Apolipoprotein A-1 protected hepatic ischaemia-reperfusion injury through suppressing macrophage pyroptosis via TLR4-NF-kappaB pathway. Liver Int. 2023, 43, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Mineo, C.; Shaul, P.W. Regulation of signal transduction by HDL. J. Lipid Res. 2013, 54, 2315–2324. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Kitano, H. A comprehensive map of the toll-like receptor signaling network. Mol. Syst. Biol. 2006, 2, 2006.0015. [Google Scholar] [CrossRef] [PubMed]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Miron, J.; Picard, C.; Frappier, J.; Dea, D.; Theroux, L.; Poirier, J. TLR4 Gene Expression and Pro-Inflammatory Cytokines in Alzheimer’s Disease and in Response to Hippocampal Deafferentation in Rodents. J. Alzheimers Dis. 2018, 63, 1547–1556. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schuffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef]
- Gorecki, A.M.; Anyaegbu, C.C.; Anderton, R.S. TLR2 and TLR4 in Parkinson’s disease pathogenesis: The environment takes a toll on the gut. Transl. Neurodegener. 2021, 10, 47. [Google Scholar] [CrossRef]
- Asadzadeh Manjili, F.; Yousefi-Ahmadipour, A.; Kazemi Arababadi, M. The roles played by TLR4 in the pathogenesis of multiple sclerosis; A systematic review article. Immunol. Lett. 2020, 220, 63–70. [Google Scholar] [CrossRef]
- Trotta, T.; Porro, C.; Calvello, R.; Panaro, M.A. Biological role of Toll-like receptor-4 in the brain. J. Neuroimmunol. 2014, 268, 1–12. [Google Scholar] [CrossRef]
- Rosenzweig, H.L.; Lessov, N.S.; Henshall, D.C.; Minami, M.; Simon, R.P.; Stenzel-Poore, M.P. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke 2004, 35, 2576–2581. [Google Scholar] [CrossRef]
- Franco, P.J.; Fernandez, D.C.; Sande, P.H.; Keller Sarmiento, M.I.; Chianelli, M.; Saenz, D.A.; Rosenstein, R.E. Effect of bacterial lipopolysaccharide on ischemic damage in the rat retina. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4604–4612. [Google Scholar] [CrossRef]
- Yi, H.; Patel, A.K.; Sodhi, C.P.; Hackam, D.J.; Hackam, A.S. Novel role for the innate immune receptor Toll-like receptor 4 (TLR4) in the regulation of the Wnt signaling pathway and photoreceptor apoptosis. PLoS ONE 2012, 7, e36560. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Chen, Y.; Kevany, B.M.; Pearlman, E.; Miyagi, M.; Maeda, T.; Palczewski, K.; Maeda, A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J. Biol. Chem. 2013, 288, 15326–15341. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhou, T.; Sun, X.; Zheng, Y.; Cheng, B.; Li, M.; Liu, X.; He, C. Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation. Cell Death Differ. 2018, 25, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Bohm, M.R.; Schallenberg, M.; Brockhaus, K.; Melkonyan, H.; Thanos, S. The pro-inflammatory role of high-mobility group box 1 protein (HMGB-1) in photoreceptors and retinal explants exposed to elevated pressure. Lab. Investig. 2016, 96, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Chen, X.; Chen, S.; Luo, Q.; Liu, X.; He, A.; He, S.; Qiu, J.; Chen, P.; Wu, Y.; et al. Tetramethylpyrazine attenuates endotoxin-induced retinal inflammation by inhibiting microglial activation via the TLR4/NF-kappaB signalling pathway. Biomed. Pharmacother. 2020, 128, 110273. [Google Scholar] [CrossRef] [PubMed]
- Chi, W.; Li, F.; Chen, H.; Wang, Y.; Zhu, Y.; Yang, X.; Zhu, J.; Wu, F.; Ouyang, H.; Ge, J.; et al. Caspase-8 promotes NLRP1/NLRP3 inflammasome activation and IL-1beta production in acute glaucoma. Proc. Natl. Acad. Sci. USA 2014, 111, 11181–11186. [Google Scholar] [CrossRef]
- Luo, C.; Yang, X.; Kain, A.D.; Powell, D.W.; Kuehn, M.H.; Tezel, G. Glaucomatous tissue stress and the regulation of immune response through glial Toll-like receptor signaling. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5697–5707. [Google Scholar] [CrossRef]
- Navarro-Partida, J.; Martinez-Rizo, A.B.; Ramirez-Barrera, P.; Velazquez-Fernandez, J.B.; Mondragon-Jaimes, V.A.; Santos-Garcia, A.; Benites-Godinez, V. Association of Toll-like receptor 4 single-nucleotide polymorphisms Asp299Gly and Thr399Ile with the risk of primary open angle glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 995–1001. [Google Scholar] [CrossRef]
- Shibuya, E.; Meguro, A.; Ota, M.; Kashiwagi, K.; Mabuchi, F.; Iijima, H.; Kawase, K.; Yamamoto, T.; Nakamura, M.; Negi, A.; et al. Association of Toll-like receptor 4 gene polymorphisms with normal tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4453–4457. [Google Scholar] [CrossRef]
- Kim, K.Y.; Perkins, G.A.; Shim, M.S.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.K. DRP1 inhibition rescues retinal ganglion cells and their axons by preserving mitochondrial integrity in a mouse model of glaucoma. Cell Death Dis. 2015, 6, e1839. [Google Scholar] [CrossRef]
- Ju, W.K.; Kim, K.Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased mitochondrial fission and volume density by blocking glutamate excitotoxicity protect glaucomatous optic nerve head astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Inman, D.M.; Steele, M.R.; Wu, G.; Soto, I.; Marsh-Armstrong, N.; Hubbard, W.C.; Calkins, D.J.; Horner, P.J.; Vetter, M.L. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Rojas, B.; Gallego, B.I.; Ramirez, A.I.; Salazar, J.J.; de Hoz, R.; Valiente-Soriano, F.J.; Aviles-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Trivino, A.; et al. Microglia in mouse retina contralateral to experimental glaucoma exhibit multiple signs of activation in all retinal layers. J. Neuroinflamm. 2014, 11, 133. [Google Scholar] [CrossRef]
- Bosco, A.; Crish, S.D.; Steele, M.R.; Romero, C.O.; Inman, D.M.; Horner, P.J.; Calkins, D.J.; Vetter, M.L. Early reduction of microglia activation by irradiation in a model of chronic glaucoma. PLoS ONE 2012, 7, e43602. [Google Scholar] [CrossRef] [PubMed]
- Bosco, A.; Steele, M.R.; Vetter, M.L. Early microglia activation in a mouse model of chronic glaucoma. J. Comp. Neurol. 2011, 519, 599–620. [Google Scholar] [CrossRef]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 2022, 87, 100998. [Google Scholar] [CrossRef] [PubMed]
- John, S.W.; Smith, R.S.; Savinova, O.V.; Hawes, N.L.; Chang, B.; Turnbull, D.; Davisson, M.; Roderick, T.H.; Heckenlively, J.R. Essential iris atrophy, pigment dispersion, and glaucoma in DBA/2J mice. Investig. Ophthalmol. Vis. Sci. 1998, 39, 951–962. [Google Scholar]
- Anderson, M.G.; Smith, R.S.; Hawes, N.L.; Zabaleta, A.; Chang, B.; Wiggs, J.L.; John, S.W. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet. 2002, 30, 81–85. [Google Scholar] [CrossRef]
- Bosco, A.; Romero, C.O.; Breen, K.T.; Chagovetz, A.A.; Steele, M.R.; Ambati, B.K.; Vetter, M.L. Neurodegeneration severity can be predicted from early microglia alterations monitored in vivo in a mouse model of chronic glaucoma. Dis. Model. Mech. 2015, 8, 443–455. [Google Scholar] [CrossRef]
- Wang, K.; Peng, B.; Lin, B. Fractalkine receptor regulates microglial neurotoxicity in an experimental mouse glaucoma model. Glia 2014, 62, 1943–1954. [Google Scholar] [CrossRef]
- Roh, M.; Zhang, Y.; Murakami, Y.; Thanos, A.; Lee, S.C.; Vavvas, D.G.; Benowitz, L.I.; Miller, J.W. Etanercept, a widely used inhibitor of tumor necrosis factor-alpha (TNF-alpha), prevents retinal ganglion cell loss in a rat model of glaucoma. PLoS ONE 2012, 7, e40065. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor necrosis factor-alpha mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [PubMed]
- Sapienza, A.; Raveu, A.L.; Reboussin, E.; Roubeix, C.; Boucher, C.; Degardin, J.; Godefroy, D.; Rostene, W.; Reaux-Le Goazigo, A.; Baudouin, C.; et al. Bilateral neuroinflammatory processes in visual pathways induced by unilateral ocular hypertension in the rat. J. Neuroinflamm. 2016, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Naskar, R.; Wissing, M.; Thanos, S. Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2962–2968. [Google Scholar]
- Johnson, E.C.; Jia, L.; Cepurna, W.O.; Doser, T.A.; Morrison, J.C. Global changes in optic nerve head gene expression after exposure to elevated intraocular pressure in a rat glaucoma model. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3161–3177. [Google Scholar] [CrossRef]
- Lozano, D.C.; Choe, T.E.; Cepurna, W.O.; Morrison, J.C.; Johnson, E.C. Early Optic Nerve Head Glial Proliferation and Jak-Stat Pathway Activation in Chronic Experimental Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 921–932. [Google Scholar] [CrossRef]
- Oikawa, K.; Ver Hoeve, J.N.; Teixeira, L.B.C.; Snyder, K.C.; Kiland, J.A.; Ellinwood, N.M.; McLellan, G.J. Sub-region-Specific Optic Nerve Head Glial Activation in Glaucoma. Mol. Neurobiol. 2020, 57, 2620–2638. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Niwa, M.; Aoki, H.; Hirata, A.; Tomita, H.; Green, P.G.; Hara, A. Retinal Cell Degeneration in Animal Models. Int. J. Mol. Sci. 2016, 17, 110. [Google Scholar] [CrossRef]
- Lin, S.; Liang, Y.; Zhang, J.; Bian, C.; Zhou, H.; Guo, Q.; Xiong, Y.; Li, S.; Su, B. Microglial TIR-domain-containing adapter-inducing interferon-beta (TRIF) deficiency promotes retinal ganglion cell survival and axon regeneration via nuclear factor-kappaB. J. Neuroinflamm. 2012, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Ueda, H. Prothymosin-alpha preconditioning activates TLR4-TRIF signaling to induce protection of ischemic retina. J. Neurochem. 2015, 135, 1161–1177. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, B.; Hu, Y.; Lu, L.; Lu, X.; Wang, J.; Xu, F.; Yu, S.; Huang, J.; Liang, X. Wogonin prevents TLR4-NF-kappaB-medicated neuro-inflammation and improves retinal ganglion cells survival in retina after optic nerve crush. Oncotarget 2016, 7, 72503–72517. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Shimazawa, M.; Ojino, K.; Izawa, H.; Takeuchi, H.; Inoue, Y.; Tsuruma, K.; Hara, H. Toll-like receptor 4 inhibitor protects against retinal ganglion cell damage induced by optic nerve crush in mice. J. Pharmacol. Sci. 2017, 133, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.; Kocab, A.J.; Zacks, D.N.; Marshak-Rothstein, A.; Gregory-Ksander, M. A small peptide antagonist of the Fas receptor inhibits neuroinflammation and prevents axon degeneration and retinal ganglion cell death in an inducible mouse model of glaucoma. J. Neuroinflamm. 2019, 16, 184. [Google Scholar] [CrossRef] [PubMed]
- Astafurov, K.; Elhawy, E.; Ren, L.; Dong, C.Q.; Igboin, C.; Hyman, L.; Griffen, A.; Mittag, T.; Danias, J. Oral microbiome link to neurodegeneration in glaucoma. PLoS ONE 2014, 9, e104416. [Google Scholar] [CrossRef] [PubMed]
- Bohush, A.; Niewiadomska, G.; Filipek, A. Role of Mitogen Activated Protein Kinase Signaling in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 2973. [Google Scholar] [CrossRef]
- Sawe, N.; Steinberg, G.; Zhao, H. Dual roles of the MAPK/ERK1/2 cell signaling pathway after stroke. J. Neurosci. Res. 2008, 86, 1659–1669. [Google Scholar] [CrossRef]
- Yu, Q.; Du, F.; Douglas, J.T.; Yu, H.; Yan, S.S.; Yan, S.F. Mitochondrial Dysfunction Triggers Synaptic Deficits via Activation of p38 MAP Kinase Signaling in Differentiated Alzheimer’s Disease Trans-Mitochondrial Cybrid Cells. J. Alzheimers Dis. 2017, 59, 223–239. [Google Scholar] [CrossRef]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef]
- Kang, Y.J.; Chen, J.; Otsuka, M.; Mols, J.; Ren, S.; Wang, Y.; Han, J. Macrophage deletion of p38alpha partially impairs lipopolysaccharide-induced cellular activation. J. Immunol. 2008, 180, 5075–5082. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kang, Y.J.; Ren, J.; Jiang, H.; Wang, Y.; Omata, M.; Han, J. Distinct effects of p38alpha deletion in myeloid lineage and gut epithelia in mouse models of inflammatory bowel disease. Gastroenterology 2010, 138, 1255–1265. [Google Scholar] [CrossRef]
- Fang, L.; Miller, Y.I. Regulation of lipid rafts, angiogenesis and inflammation by AIBP. Curr. Opin. Lipidol. 2019, 30, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Lane, N. Mitochondrial disease: Powerhouse of disease. Nature 2006, 440, 600–602. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef] [PubMed]
- Ju, W.K.; Kim, K.Y.; Lindsey, J.D.; Angert, M.; Duong-Polk, K.X.; Scott, R.T.; Kim, J.J.; Kukhmazov, I.; Ellisman, M.H.; Perkins, G.A.; et al. Intraocular pressure elevation induces mitochondrial fission and triggers OPA1 release in glaucomatous optic nerve. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4903–4911. [Google Scholar] [CrossRef] [PubMed]
- Marbaix, A.Y.; Tyteca, D.; Niehaus, T.D.; Hanson, A.D.; Linster, C.L.; Van Schaftingen, E. Occurrence and subcellular distribution of the NADPHX repair system in mammals. Biochem. J. 2014, 460, 49–58. [Google Scholar] [CrossRef]
- Duan, M.; Chen, H.; Yin, L.; Zhu, X.; Novak, P.; Lv, Y.; Zhao, G.; Yin, K. Mitochondrial apolipoprotein A-I binding protein alleviates atherosclerosis by regulating mitophagy and macrophage polarization. Cell Commun. Signal 2022, 20, 60. [Google Scholar] [CrossRef]
- Solsona-Vilarrasa, E.; Fucho, R.; Torres, S.; Nunez, S.; Nuno-Lambarri, N.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol enrichment in liver mitochondria impairs oxidative phosphorylation and disrupts the assembly of respiratory supercomplexes. Redox Biol. 2019, 24, 101214. [Google Scholar] [CrossRef]
- Torres, S.; Garcia-Ruiz, C.M.; Fernandez-Checa, J.C. Mitochondrial Cholesterol in Alzheimer’s Disease and Niemann-Pick Type C Disease. Front. Neurol. 2019, 10, 1168. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Huang, P.; Yu, H.; Chen, J.; Liu, X.; Wang, J.; Shen, X.; Zhong, Y. Hydrogen sulfide supplement preserves mitochondrial function of retinal ganglion cell in a rat glaucoma model. Cell Tissue Res. 2022, 389, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Tok, L.; Naziroglu, M.; Uguz, A.C.; Tok, O. Elevated hydrostatic pressures induce apoptosis and oxidative stress through mitochondrial membrane depolarization in PC12 neuronal cells: A cell culture model of glaucoma. J. Recept. Signal Transduct. Res. 2014, 34, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Harun-Or-Rashid, M.; Inman, D.M. Reduced AMPK activation and increased HCAR activation drive anti-inflammatory response and neuroprotection in glaucoma. J. Neuroinflamm. 2018, 15, 313. [Google Scholar] [CrossRef]
- Chen, S.; Wang, N.; Xiong, S.; Xia, X. The correlation between primary open-angle glaucoma (POAG) and gut microbiota: A pilot study towards predictive, preventive, and personalized medicine. EPMA J. 2023, 14, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yu, N.; Ye, Z.; Gu, Y.; Zhang, C.; Chen, M.; Wang, K. Inhibition of cGAS-STING pathway alleviates neuroinflammation-induced retinal ganglion cell death after ischemia/reperfusion injury. Cell Death Dis. 2023, 14, 615. [Google Scholar] [CrossRef] [PubMed]
- Baudouin, C.; Kolko, M.; Melik-Parsadaniantz, S.; Messmer, E.M. Inflammation in Glaucoma: From the back to the front of the eye, and beyond. Prog. Retin. Eye Res. 2021, 83, 100916. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- Linster, C.L.; Van Schaftingen, E.; Hanson, A.D. Metabolite damage and its repair or pre-emption. Nat. Chem. Biol. 2013, 9, 72–80. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Manor, J.; Calame, D.; Gijavanekar, C.; Fisher, K.; Hunter, J.; Mizerik, E.; Bacino, C.; Scaglia, F.; Elsea, S.H. NAXE deficiency: A neurometabolic disorder of NAD(P)HX repair amenable for metabolic correction. Mol. Genet. Metab. 2022, 136, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Marbaix, A.Y.; Noel, G.; Detroux, A.M.; Vertommen, D.; Van Schaftingen, E.; Linster, C.L. Extremely conserved ATP- or ADP-dependent enzymatic system for nicotinamide nucleotide repair. J. Biol. Chem. 2011, 286, 41246–41252. [Google Scholar] [CrossRef] [PubMed]
- Shumilin, I.A.; Cymborowski, M.; Chertihin, O.; Jha, K.N.; Herr, J.C.; Lesley, S.A.; Joachimiak, A.; Minor, W. Identification of unknown protein function using metabolite cocktail screening. Structure 2012, 20, 1715–1725. [Google Scholar] [CrossRef]
- Rafter, G.W.; Chaykin, S.; Krebs, E.G. The action of glyceraldehyde-3-phosphate dehydrogenase on reduced diphosphopyridine nucleotide. J. Biol. Chem. 1954, 208, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Dave, V. Inhibition of NADP-dependent dehydrogenases by modified products of NADPH. Arch. Biochem. Biophys. 1975, 169, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Meng, S.; Gu, Q.; Araujo-Gutierrez, R.; Kumar, S.; Yan, Q.; Almazan, F.; Youker, K.A.; Fu, Y.; Pownall, H.J.; et al. AIBP Limits Angiogenesis Through gamma-Secretase-Mediated Upregulation of Notch Signaling. Circ. Res. 2017, 120, 1727–1739. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W. Vitamin B(3) modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef]
- Hui, F.; Tang, J.; Williams, P.A.; McGuinness, M.B.; Hadoux, X.; Casson, R.J.; Coote, M.; Trounce, I.A.; Martin, K.R.; van Wijngaarden, P.; et al. Improvement in inner retinal function in glaucoma with nicotinamide (vitamin B3) supplementation: A crossover randomized clinical trial. Clin. Exp. Ophthalmol. 2020, 48, 903–914. [Google Scholar] [CrossRef]
- Kouassi Nzoughet, J.; Chao de la Barca, J.M.; Guehlouz, K.; Leruez, S.; Coulbault, L.; Allouche, S.; Bocca, C.; Muller, J.; Amati-Bonneau, P.; Gohier, P.; et al. Nicotinamide Deficiency in Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2509–2514. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e1065. [Google Scholar] [CrossRef]
- Kim, J.D.; Zhu, L.; Sun, Q.; Fang, L. Systemic metabolite profiling reveals sexual dimorphism of AIBP control of metabolism in mice. PLoS ONE 2021, 16, e0248964. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.Q.; Wang, Y.S. The role of Toll-like receptors in retinal ischemic diseases. Int. J. Ophthalmol. 2016, 9, 1343–1351. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.; Choi, S.-H.; Bastola, T.; Park, Y.; Oh, J.; Kim, K.-Y.; Hwang, S.; Miller, Y.I.; Ju, W.-K. AIBP: A New Safeguard against Glaucomatous Neuroinflammation. Cells 2024, 13, 198. https://doi.org/10.3390/cells13020198
Choi S, Choi S-H, Bastola T, Park Y, Oh J, Kim K-Y, Hwang S, Miller YI, Ju W-K. AIBP: A New Safeguard against Glaucomatous Neuroinflammation. Cells. 2024; 13(2):198. https://doi.org/10.3390/cells13020198
Chicago/Turabian StyleChoi, Seunghwan, Soo-Ho Choi, Tonking Bastola, Younggun Park, Jonghyun Oh, Keun-Young Kim, Sinwoo Hwang, Yury I. Miller, and Won-Kyu Ju. 2024. "AIBP: A New Safeguard against Glaucomatous Neuroinflammation" Cells 13, no. 2: 198. https://doi.org/10.3390/cells13020198
APA StyleChoi, S., Choi, S. -H., Bastola, T., Park, Y., Oh, J., Kim, K. -Y., Hwang, S., Miller, Y. I., & Ju, W. -K. (2024). AIBP: A New Safeguard against Glaucomatous Neuroinflammation. Cells, 13(2), 198. https://doi.org/10.3390/cells13020198