
Perspective Strategies for Interventions in Parkinsonism: Remedying the Neglected Role of TPPP


Abstract
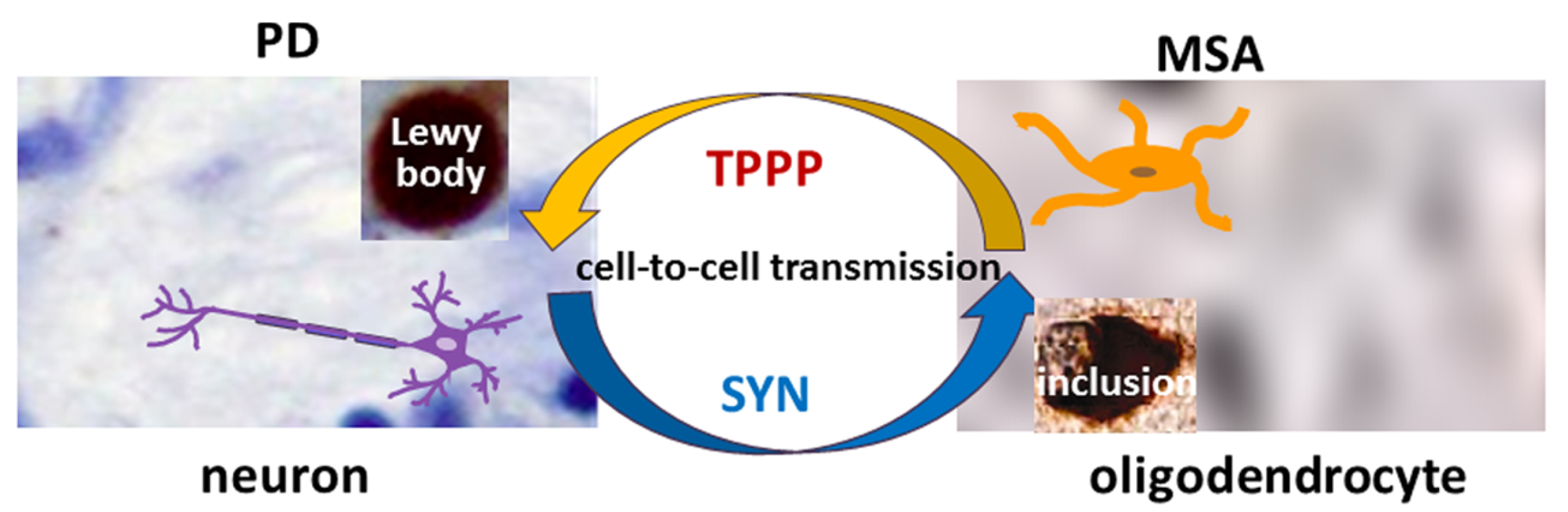
:1. Introduction
2. Pathological Interaction between TPPP and SYN
3. The Role of TPPP in the Pathomechanism of Synucleinopathies: Protein Aggregation
4. The Role of TPPP in the Dysregulation of Protein Degradation in Parkinsonism
5. SYN and TPPP as Biomarkers
6. Targeting the Interface of the Pathological SYN-TPPP Complex
7. PROTAC and Related Technologies for the Elimination of Unwanted Proteins
8. Targeting mRNA in Parkinsonism
9. Conclusions and Beyond
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Delvadia, P.; Joharapurkar, A.; Shah, J. Uncovering Novel Therapeutic Targets for Parkinson’s Disease. ACS Chem. Neurosci. 2023, 14, 1935–1949. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Yoshimoto, M.; Tsuji, S.; Takahashi, H. Alpha-synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci. Lett. 1998, 249, 180–182. [Google Scholar] [CrossRef]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Graves, N.J.; Gambin, Y.; Sierecki, E. alpha-Synuclein Strains and Their Relevance to Parkinson’s Disease, Multiple System Atrophy, and Dementia with Lewy Bodies. Int. J. Mol. Sci. 2023, 24, 12134. [Google Scholar] [CrossRef]
- Stefanova, N.; Wenning, G.K. Multiple system atrophy: At the crossroads of cellular, molecular and genetic mechanisms. Nat. Rev. Neurosci. 2023, 24, 334–346. [Google Scholar] [CrossRef]
- Hill, D.R.; Huters, A.D.; Towne, T.B.; Reddy, R.E.; Fogle, J.L.; Voight, E.A.; Kym, P.R. Parkinson’s Disease: Advances in Treatment and the Syntheses of Various Classes of Pharmaceutical Drug Substances. Chem. Rev. 2023, 123, 13693–13712. [Google Scholar] [CrossRef]
- Stott, S.R.W.; Wyse, R.K.; Brundin, P. Novel approaches to counter protein aggregation pathology in Parkinson’s disease. Prog. Brain Res. 2020, 252, 451–492. [Google Scholar]
- Morris, H.R.; Spillantini, M.G.; Sue, C.M.; Williams-Gray, C.H. The pathogenesis of Parkinson’s disease. Lancet 2024, 403, 293–304. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Laszlo, L.; Kovacs, J.; Jensen, P.H.; Lindersson, E.; Botond, G.; Molnar, T.; Perczel, A.; Hudecz, F.; Mezo, G.; et al. Natively unfolded tubulin polymerization promoting protein TPPP/p25 is a common marker of alpha-synucleinopathies. Neurobiol. Dis. 2004, 17, 155–162. [Google Scholar] [CrossRef]
- Olah, J.; Lehotzky, A.; Szunyogh, S.; Szenasi, T.; Orosz, F.; Ovadi, J. Microtubule-Associated Proteins with Regulatory Functions by Day and Pathological Potency at Night. Cells 2020, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidi, P.; Arvanitaki, F.; Karakitsou, A.K.; Vetsi, M.; Kloukina, I.; Zweckstetter, M.; Giller, K.; Becker, S.; Sorrentino, Z.A.; Giasson, B.I.; et al. Endogenous oligodendroglial alpha-synuclein and TPPP/p25alpha orchestrate alpha-synuclein pathology in experimental multiple system atrophy models. Acta Neuropathol. 2019, 138, 415–441. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Gelpi, E.; Lehotzky, A.; Hoftberger, R.; Erdei, A.; Budka, H.; Ovadi, J. The brain-specific protein TPPP/p25 in pathological protein deposits of neurodegenerative diseases. Acta Neuropathol. 2007, 113, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Tokesi, N.; Olah, J.; Hlavanda, E.; Szunyogh, S.; Szabo, A.; Babos, F.; Magyar, A.; Lehotzky, A.; Vass, E.; Ovadi, J. Identification of motives mediating alternative functions of the neomorphic moonlighting TPPP/p25. Biochim. Biophys. Acta 2014, 1842, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Lehotzky, A.; Lau, P.; Tokesi, N.; Muja, N.; Hudson, L.D.; Ovadi, J. Tubulin polymerization-promoting protein (TPPP/p25) is critical for oligodendrocyte differentiation. Glia 2010, 58, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Gim, Y.; Jang, E.H.; Hur, E.M. Functions and dysfunctions of oligodendrocytes in neurodegenerative diseases. Front. Cell. Neurosci. 2022, 16, 1083159. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Obayashi, M.; Ozaki, K.; Ichinose, S.; Kakita, A.; Tada, M.; Takahashi, H.; Ando, N.; Eishi, Y.; Mizusawa, H.; et al. Relocation of p25alpha/tubulin polymerization promoting protein from the nucleus to the perinuclear cytoplasm in the oligodendroglia of sporadic and COQ2 mutant multiple system atrophy. Acta Neuropathol. Commun. 2014, 2, 136. [Google Scholar] [PubMed]
- Tokesi, N.; Lehotzky, A.; Horvath, I.; Szabo, B.; Olah, J.; Lau, P.; Ovadi, J. TPPP/p25 promotes tubulin acetylation by inhibiting histone deacetylase 6. J. Biol. Chem. 2010, 285, 17896–17906. [Google Scholar] [CrossRef]
- Szabo, A.; Olah, J.; Szunyogh, S.; Lehotzky, A.; Szenasi, T.; Csaplar, M.; Schiedel, M.; Low, P.; Jung, M.; Ovadi, J. Modulation of Microtubule Acetylation by the Interplay Of TPPP/p25, SIRT2 and New Anticancer Agents with Anti-SIRT2 Potency. Sci. Rep. 2017, 7, 17070. [Google Scholar] [CrossRef] [PubMed]
- Olah, J.; Szenasi, T.; Szunyogh, S.; Szabo, A.; Lehotzky, A.; Ovadi, J. Further evidence for microtubule-independent dimerization of TPPP/p25. Sci. Rep. 2017, 7, 40594. [Google Scholar] [CrossRef] [PubMed]
- Zotter, A.; Bodor, A.; Olah, J.; Hlavanda, E.; Orosz, F.; Perczel, A.; Ovadi, J. Disordered TPPP/p25 binds GTP and displays Mg2+-dependent GTPase activity. FEBS Lett. 2011, 585, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Zotter, A.; Olah, J.; Hlavanda, E.; Bodor, A.; Perczel, A.; Szigeti, K.; Fidy, J.; Ovadi, J. Zn(2)+-induced rearrangement of the disordered TPPP/p25 affects its microtubule assembly and GTPase activity. Biochemistry 2011, 50, 9568–9578. [Google Scholar] [CrossRef]
- Tirian, L.; Hlavanda, E.; Olah, J.; Horvath, I.; Orosz, F.; Szabo, B.; Kovacs, J.; Szabad, J.; Ovadi, J. TPPP/p25 promotes tubulin assemblies and blocks mitotic spindle formation. Proc. Natl. Acad. Sci. USA 2003, 100, 13976–13981. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Lehotzky, A.; Budka, H.; Ovadi, J.; Kovacs, G.G. TPPP/p25 in brain tumours: Expression in non-neoplastic oligodendrocytes but not in oligodendroglioma cells. Acta Neuropathol. 2007, 113, 213–215. [Google Scholar] [CrossRef]
- Xie, J.; Chen, S.; Bopassa, J.C.; Banerjee, S. Drosophila tubulin polymerization promoting protein mutants reveal pathological correlates relevant to human Parkinson’s disease. Sci. Rep. 2021, 11, 13614. [Google Scholar] [CrossRef]
- Barbato, E.; Darrah, R.; Kelley, T.J. Tubulin Polymerization Promoting Protein Affects the Circadian Timing System in C57Bl/6 Mice. J. Circadian Rhythm. 2021, 19, 5. [Google Scholar] [CrossRef]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papic, E.; Racki, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. Off. J. Soc. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Bates, C.A.; Zheng, W. Brain disposition of alpha-Synuclein: Roles of brain barrier systems and implications for Parkinson’s disease. Fluids Barriers CNS 2014, 11, 17. [Google Scholar] [CrossRef]
- Takahashi, M.; Tomizawa, K.; Fujita, S.C.; Sato, K.; Uchida, T.; Imahori, K. A brain-specific protein p25 is localized and associated with oligodendrocytes, neuropil, and fiber-like structures of the CA hippocampal region in the rat brain. J. Neurochem. 1993, 60, 228–235. [Google Scholar] [CrossRef]
- Valdinocci, D.; Radford, R.A.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular alpha-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, 469. [Google Scholar] [CrossRef]
- Bras, I.C.; Outeiro, T.F. Alpha-Synuclein: Mechanisms of Release and Pathology Progression in Synucleinopathies. Cells 2021, 10, 375. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Han, C.; Ma, K.; Guo, X.; Wan, F.; Kou, L.; Yin, S.; Liu, L.; Huang, J.; et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Szenasi, T.; Olah, J.; Szabo, A.; Szunyogh, S.; Lang, A.; Perczel, A.; Lehotzky, A.; Uversky, V.N.; Ovadi, J. Challenging drug target for Parkinson’s disease: Pathological complex of the chameleon TPPP/p25 and alpha-synuclein proteins. Biochim. Biophys. Acta. Mol. Basis Dis. 2017, 1863, 310–323. [Google Scholar] [CrossRef] [PubMed]
- Szunyogh, S.; Olah, J.; Szenasi, T.; Szabo, A.; Ovadi, J. Targeting the interface of the pathological complex of alpha-synuclein and TPPP/p25. Biochim. Biophys. Acta 2015, 1852, 2653–2661. [Google Scholar] [CrossRef] [PubMed]
- Keerthikumar, S.; Gangoda, L.; Liem, M.; Fonseka, P.; Atukorala, I.; Ozcitti, C.; Mechler, A.; Adda, C.G.; Ang, C.S.; Mathivanan, S. Proteogenomic analysis reveals exosomes are more oncogenic than ectosomes. Oncotarget 2015, 6, 15375–15396. [Google Scholar] [CrossRef]
- Kon, T.; Forrest, S.L.; Lee, S.; Martinez-Valbuena, I.; Li, J.; Nassir, N.; Uddin, M.J.; Lang, A.E.; Kovacs, G.G. Neuronal SNCA transcription during Lewy body formation. Acta Neuropathol. Commun. 2023, 11, 185. [Google Scholar] [CrossRef]
- Gould, R.; Brady, S. Identifying mRNAs Residing in Myelinating Oligodendrocyte Processes as a Basis for Understanding Internode Autonomy. Life 2023, 13, 945. [Google Scholar] [CrossRef]
- Cui, H.; Kilpelainen, T.; Zouzoula, L.; Auno, S.; Trontti, K.; Kurvonen, S.; Norrbacka, S.; Hovatta, I.; Jensen, P.H.; Myohanen, T.T. Prolyl oligopeptidase inhibition reduces alpha-synuclein aggregation in a cellular model of multiple system atrophy. J. Cell. Mol. Med. 2021, 25, 9634–9646. [Google Scholar] [CrossRef]
- Kaji, S.; Maki, T.; Kinoshita, H.; Uemura, N.; Ayaki, T.; Kawamoto, Y.; Furuta, T.; Urushitani, M.; Hasegawa, M.; Kinoshita, Y.; et al. Pathological Endogenous alpha-Synuclein Accumulation in Oligodendrocyte Precursor Cells Potentially Induces Inclusions in Multiple System Atrophy. Stem Cell Rep. 2018, 10, 356–365. [Google Scholar] [CrossRef]
- Ferreira, N.; Gram, H.; Sorrentino, Z.A.; Gregersen, E.; Schmidt, S.I.; Reimer, L.; Betzer, C.; Perez-Gozalbo, C.; Beltoja, M.; Nagaraj, M.; et al. Multiple system atrophy-associated oligodendroglial protein p25alpha stimulates formation of novel alpha-synuclein strain with enhanced neurodegenerative potential. Acta Neuropathol. 2021, 142, 87–115. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Sakunthala, A.; Gadhe, L.; Poudyal, M.; Sawner, A.S.; Kadu, P.; Maji, S.K. Liquid-liquid Phase Separation of alpha-Synuclein: A New Mechanistic Insight for alpha-Synuclein Aggregation Associated with Parkinson’s Disease Pathogenesis. J. Mol. Biol. 2023, 435, 167713. [Google Scholar] [CrossRef] [PubMed]
- Rohan, Z.; Milenkovic, I.; Lutz, M.I.; Matej, R.; Kovacs, G.G. Shared and Distinct Patterns of Oligodendroglial Response in alpha-Synucleinopathies and Tauopathies. J. Neuropathol. Exp. Neurol. 2016, 75, 1100–1109. [Google Scholar] [CrossRef] [PubMed]
- De Nuccio, F.; Kashyrina, M.; Serinelli, F.; Laferriere, F.; Lofrumento, D.D.; De Giorgi, F.; Ichas, F. Oligodendrocytes Prune Axons Containing alpha-Synuclein Aggregates In Vivo: Lewy Neurites as Precursors of Glial Cytoplasmic Inclusions in Multiple System Atrophy? Biomolecules 2023, 13, 269. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Masaki, K.; Matsuse, D.; Yamaguchi, H.; Tanaka, T.; Matsuo, E.; Hayashida, S.; Watanabe, M.; Matsushita, T.; Sadashima, S.; et al. Early and extensive alterations of glial connexins, distal oligodendrogliopathy type demyelination, and nodal/paranodal pathology are characteristic of multiple system atrophy. Brain Pathol. 2023, 33, e13131. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Baba, T.; Kobayashi, M.; Konno, M.; Sugeno, N.; Kikuchi, A.; Itoyama, Y.; Takeda, A. Role of TPPP/p25 on alpha-synuclein-mediated oligodendroglial degeneration and the protective effect of SIRT2 inhibition in a cellular model of multiple system atrophy. Neurochem. Int. 2010, 57, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Lehotzky, A.; Olah, J.; Fekete, J.T.; Szenasi, T.; Szabo, E.; Gyorffy, B.; Varady, G.; Ovadi, J. Co-Transmission of Alpha-Synuclein and TPPP/p25 Inhibits Their Proteolytic Degradation in Human Cell Models. Front. Mol. Biosci. 2021, 8, 666026. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mirasierra, I.; Ghimire, S.; Hernandez-Diaz, S.; Soukup, S.F. Targeting Macroautophagy as a Therapeutic Opportunity to Treat Parkinson’s Disease. Front. Cell Dev. Biol. 2022, 10, 921314. [Google Scholar] [CrossRef] [PubMed]
- Mavroeidi, P.; Arvanitaki, F.; Vetsi, M.; Becker, S.; Vlachakis, D.; Jensen, P.H.; Stefanis, L.; Xilouri, M. Autophagy mediates the clearance of oligodendroglial SNCA/alpha-synuclein and TPPP/p25A in multiple system atrophy models. Autophagy 2022, 18, 2104–2133. [Google Scholar] [CrossRef]
- Borland, H.; Rasmussen, I.; Bjerregaard-Andersen, K.; Rasmussen, M.; Olsen, A.; Vilhardt, F. alpha-synuclein buildup is alleviated via ESCRT-dependent endosomal degradation brought about by p38MAPK inhibition in cells expressing p25alpha. J. Biol. Chem. 2022, 298, 102531. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Constantinescu, J.; Nellgard, B.; Jakobsson, P.; Brum, W.S.; Gobom, J.; Forsgren, L.; Dalla, K.; Constantinescu, R.; Zetterberg, H.; et al. Cerebrospinal Fluid Biomarkers of Synaptic Dysfunction are Altered in Parkinson’s Disease and Related Disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Vendel, E.; Rottschafer, V.; de Lange, E.C.M. The need for mathematical modelling of spatial drug distribution within the brain. Fluids Barriers CNS 2019, 16, 12. [Google Scholar] [CrossRef]
- Siderowf, A.; Concha-Marambio, L.; Lafontant, D.E.; Farris, C.M.; Ma, Y.; Urenia, P.A.; Nguyen, H.; Alcalay, R.N.; Chahine, L.M.; Foroud, T.; et al. Parkinson’s Progression Markers, I. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using alpha-synuclein seed amplification: A cross-sectional study. Lancet. Neurol. 2023, 22, 407–417. [Google Scholar] [CrossRef]
- Kwon, D.H.; Hwang, J.S.; Kim, S.G.; Jang, Y.E.; Shin, T.H.; Lee, G. Cerebrospinal Fluid Metabolome in Parkinson’s Disease and Multiple System Atrophy. Int. J. Mol. Sci. 2022, 23, 1879. [Google Scholar] [CrossRef]
- Schulz, I.; Kruse, N.; Gera, R.G.; Kremer, T.; Cedarbaum, J.; Barbour, R.; Zago, W.; Schade, S.; Otte, B.; Bartl, M.; et al. Systematic Assessment of 10 Biomarker Candidates Focusing on alpha-Synuclein-Related Disorders. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 2874–2887. [Google Scholar] [CrossRef]
- Mavroudis, I.; Petridis, F.; Chatzikonstantinou, S.; Kazis, D. Alpha-synuclein Levels in the Differential Diagnosis of Lewy Bodies Dementia and Other Neurodegenerative Disorders: A Meta-analysis. Alzheimer Dis. Assoc. Disord. 2020, 34, 220–224. [Google Scholar] [CrossRef]
- Zhou, B.; Wen, M.; Yu, W.F.; Zhang, C.L.; Jiao, L. The Diagnostic and Differential Diagnosis Utility of Cerebrospinal Fluid alpha-Synuclein Levels in Parkinson’s Disease: A Meta-Analysis. Park. Dis. 2015, 2015, 567386. [Google Scholar]
- Xiang, C.; Cong, S.; Tan, X.; Ma, S.; Liu, Y.; Wang, H.; Cong, S. A meta-analysis of the diagnostic utility of biomarkers in cerebrospinal fluid in Parkinson’s disease. NPJ Park. Dis. 2022, 8, 165. [Google Scholar] [CrossRef]
- Hoftberger, R.; Fink, S.; Aboul-Enein, F.; Botond, G.; Olah, J.; Berki, T.; Ovadi, J.; Lassmann, H.; Budka, H.; Kovacs, G.G. Tubulin polymerization promoting protein (TPPP/p25) as a marker for oligodendroglial changes in multiple sclerosis. Glia 2010, 58, 1847–1857. [Google Scholar] [CrossRef]
- Vincze, O.; Olah, J.; Zadori, D.; Klivenyi, P.; Vecsei, L.; Ovadi, J. A new myelin protein, TPPP/p25, reduced in demyelinated lesions is enriched in cerebrospinal fluid of multiple sclerosis. Biochem. Biophys. Res. Commun. 2011, 409, 137–141. [Google Scholar] [CrossRef]
- Rodger, A.T.; ALNasser, M.; Carter, W.G. Are Therapies That Target alpha-Synuclein Effective at Halting Parkinson’s Disease Progression? A Systematic Review. Int. J. Mol. Sci. 2023, 24, 11022. [Google Scholar] [CrossRef]
- Pujols, J.; Pena-Diaz, S.; Lazaro, D.F.; Peccati, F.; Pinheiro, F.; Gonzalez, D.; Carija, A.; Navarro, S.; Conde-Gimenez, M.; Garcia, J.; et al. Small molecule inhibits alpha-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc. Natl. Acad. Sci. USA 2018, 115, 10481–10486. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef]
- Allen, S.G.; Meade, R.M.; White Stenner, L.L.; Mason, J.M. Peptide-based approaches to directly target alpha-synuclein in Parkinson’s disease. Mol. Neurodegener. 2023, 18, 80. [Google Scholar] [CrossRef] [PubMed]
- Nim, S.; O’Hara, D.M.; Corbi-Verge, C.; Perez-Riba, A.; Fujisawa, K.; Kapadia, M.; Chau, H.; Albanese, F.; Pawar, G.; De Snoo, M.L.; et al. Disrupting the alpha-synuclein-ESCRT interaction with a peptide inhibitor mitigates neurodegeneration in preclinical models of Parkinson’s disease. Nat. Commun. 2023, 14, 2150. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Soga, T.; Ahemad, N.; Bhuvanendran, S.; Parhar, I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from alpha-Synuclein-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2022, 23, 5193. [Google Scholar] [CrossRef]
- Kulesskaya, N.; Bhattacharjee, A.; Holmstrom, K.M.; Vuorio, P.; Henriques, A.; Callizot, N.; Huttunen, H.J. HER-096 is a CDNF-derived brain-penetrating peptidomimetic that protects dopaminergic neurons in a mouse synucleinopathy model of Parkinson’s disease. Cell Chem. Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Edwards, R.H. The physiological role of alpha-synuclein and its relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef]
- Selkoe, D.; Dettmer, U.; Luth, E.; Kim, N.; Newman, A.; Bartels, T. Defining the native state of alpha-synuclein. Neuro-Degener. Dis. 2014, 13, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Cascella, R.; Bigi, A.; Cremades, N.; Cecchi, C. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell. Mol. Life Sci. CMLS 2022, 79, 174. [Google Scholar] [CrossRef]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Crews, C.M. PROTACs: Past, present and future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Chirnomas, D.; Hornberger, K.R.; Crews, C.M. Protein degraders enter the clinic—A new approach to cancer therapy. Nat. Rev. Clin. Oncol. 2023, 20, 265–278. [Google Scholar] [CrossRef]
- Schiedel, M.; Lehotzky, A.; Szunyogh, S.; Olah, J.; Hammelmann, S.; Wossner, N.; Robaa, D.; Einsle, O.; Sippl, W.; Ovadi, J.; et al. HaloTag-Targeted Sirtuin-Rearranging Ligand (SirReal) for the Development of Proteolysis-Targeting Chimeras (PROTACs) against the Lysine Deacetylase Sirtuin 2 (Sirt2)*. Chembiochem A Eur. J. Chem. Biol. 2020, 21, 3371–3376. [Google Scholar] [CrossRef]
- Lin, X.; Xiang, H.; Luo, G. Targeting estrogen receptor alpha for degradation with PROTACs: A promising approach to overcome endocrine resistance. Eur. J. Med. Chem. 2020, 206, 112689. [Google Scholar] [CrossRef]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC Compounds Targeting alpha-Synuclein Protein for Treating Neurogenerative Disorders: Alzheimer’s and Parkinson’s Diseases. ACS Med. Chem. Lett. 2020, 11, 1086–1087. [Google Scholar] [CrossRef]
- Pedrini, M.; Iannielli, A.; Meneghelli, L.; Passarella, D.; Broccoli, V.; Seneci, P. Synthesis and Preliminary Characterization of Putative Anle138b-Centered PROTACs against alpha-Synuclein Aggregation. Pharmaceutics 2023, 15, 1467. [Google Scholar] [CrossRef]
- Wen, T.; Chen, J.; Zhang, W.; Pang, J. Design, Synthesis and Biological Evaluation of alpha-Synuclein Proteolysis-Targeting Chimeras. Molecules 2023, 28, 4458. [Google Scholar] [CrossRef]
- Tong, Y.; Zhu, W.; Chen, J.; Wen, T.; Xu, F.; Pang, J. Discovery of Small-Molecule Degraders for Alpha-Synuclein Aggregates. J. Med. Chem. 2023, 66, 7926–7942. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Ren, X.; Xue, F.; He, Y.; Zhang, R.; Zheng, Y.; Huang, H.; Wang, W.; Zhang, J. Specific Knockdown of alpha-Synuclein by Peptide-Directed Proteasome Degradation Rescued Its Associated Neurotoxicity. Cell Chem. Biol. 2020, 27, 751–762.e4. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yoon, D.; Sung, K.W.; Bae, E.J.; Park, D.H.; Suh, Y.H.; Kwon, Y.T. Targeted degradation of SNCA/alpha-synuclein aggregates in neurodegeneration using the AUTOTAC chemical platform. Autophagy 2023, 20, 463–465. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 628137. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Shin, S.H.; Lim, C.G.; Heo, Y.H.; Choi, I.Y.; Kim, H.H. Synthetic RNA Therapeutics in Cancer. J. Pharmacol. Exp. Ther. 2023, 386, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Cole, T.A.; Zhao, H.; Collier, T.J.; Sandoval, I.; Sortwell, C.E.; Steece-Collier, K.; Daley, B.F.; Booms, A.; Lipton, J.; Welch, M.; et al. alpha-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 2021, 6, e135633. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, M.A.S.; Outeiro, T.F. Epigenetics in Parkinson’s Disease. Adv. Exp. Med. Biol. 2017, 978, 363–390. [Google Scholar] [PubMed]
- Noronha, O.; Mesarosovo, L.; Anink, J.J.; Iyer, A.; Aronica, E.; Mills, J.D. Differentially Expressed miRNAs in Age-Related Neurodegenerative Diseases: A Meta-Analysis. Genes 2022, 13, 1034. [Google Scholar] [CrossRef] [PubMed]
- Juzwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef]
- Paccosi, E.; Proietti-De-Santis, L. Parkinson’s Disease: From Genetics and Epigenetics to Treatment, a miRNA-Based Strategy. Int. J. Mol. Sci. 2023, 24, 9547. [Google Scholar] [CrossRef] [PubMed]
- Szelagowski, A.; Kozakiewicz, M. A Glance at Biogenesis and Functionality of MicroRNAs and Their Role in the Neuropathogenesis of Parkinson’s Disease. Oxidative Med. Cell. Longev. 2023, 2023, 7759053. [Google Scholar] [CrossRef] [PubMed]
- Santos-Lobato, B.L.; Vidal, A.F.; Ribeiro-Dos-Santos, A. Regulatory miRNA-mRNA Networks in Parkinson’s Disease. Cells 2021, 10, 1410. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, M.; Yan, R.; Liu, J.; Maddila, S.; Junn, E.; Mouradian, M.M. MicroRNA-7 Protects Against Neurodegeneration Induced by alpha-Synuclein Preformed Fibrils in the Mouse Brain. Neurother. J. Am. Soc. Exp. NeuroTherapeutics 2021, 18, 2529–2540. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Zhang, P.; Park, H.J.; Zhang, J.; Junn, E.; Andrews, R.J.; Velagapudi, S.P.; Abegg, D.; Vishnu, K.; Costales, M.G.; Childs-Disney, J.L.; et al. Translation of the intrinsically disordered protein alpha-synuclein is inhibited by a small molecule targeting its structured mRNA. Proc. Natl. Acad. Sci. USA 2020, 117, 1457–1467. [Google Scholar] [CrossRef]
- Zhu, S.; Choudhury, N.R.; Rooney, S.; Pham, N.T.; Koszela, J.; Kelly, D.; Spanos, C.; Rappsilber, J.; Auer, M.; Michlewski, G. RNA pull-down confocal nanoscanning (RP-CONA) detects quercetin as pri-miR-7/HuR interaction inhibitor that decreases alpha-synuclein levels. Nucleic Acids Res. 2021, 49, 6456–6473. [Google Scholar] [CrossRef]
- Lopez-Cuina, M.; Guerin, P.; Dutheil, N.; Martin, C.; Lasserre, T.L.; Fernagut, P.O.; Meissner, W.G.; Bezard, E. GRK2-Targeted Knockdown as Therapy for Multiple System Atrophy. Mov. Disord. Off. J. Mov. Disord. Soc. 2023, 38, 1336–1340. [Google Scholar] [CrossRef]
- Izco, M.; Schleef, M.; Schmeer, M.; Carlos, E.; Verona, G.; Alvarez-Erviti, L. Targeted Extracellular Vesicle Gene Therapy for Modulating Alpha-Synuclein Expression in Gut and Spinal Cord. Pharmaceutics 2023, 15, 1230. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Guan, W. A comprehensive review on the importance of miRNA-206 in animal model and human diseases. Curr. Neuropharmacol. 2023, 22, 1064–1079. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.N.; Wang, Y.J.; Wang, H.; Song, L.; Chen, Y.; Wang, J.L.; Ye, Y.; Jiang, B. The Anti-dementia Effects of Donepezil Involve miR-206-3p in the Hippocampus and Cortex. Biol. Pharm. Bull. 2017, 40, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.L.; Duan, M.J.; Ma, J.C.; Xu, L.; Mao, M.; Biddyut, D.; Wang, Q.; Yang, C.; Zhang, S.; Xu, Y.; et al. Myocardial infarction-induced hippocampal microtubule damage by cardiac originating microRNA-1 in mice. J. Mol. Cell. Cardiol. 2018, 120, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Olah, J.; Krylov, S.N.; Uversky, V.N.; Ovadi, J. The Sherpa hypothesis: Phenotype-Preserving Disordered Proteins stabilize the phenotypes of neurons and oligodendrocytes. NPJ Syst. Biol. Appl. 2023, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Norris, V.; Amar, P.; Legent, G.; Ripoll, C.; Thellier, M.; Ovadi, J. Sensor potency of the moonlighting enzyme-decorated cytoskeleton: The cytoskeleton as a metabolic sensor. BMC Biochem. 2013, 14, 3. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Lee, V.M.; Smith, A.B., 3rd; Trojanowski, J.Q.; Ballatore, C. Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol. Dis. 2017, 105, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Wetzel, A.; Granno, S.; Heaton, G.; Harvey, K. Back to the tubule: Microtubule dynamics in Parkinson’s disease. Cell. Mol. Life Sci. 2017, 74, 409–434. [Google Scholar] [CrossRef]
- Fu, M.M.; McAlear, T.S.; Nguyen, H.; Oses-Prieto, J.A.; Valenzuela, A.; Shi, R.D.; Perrino, J.J.; Huang, T.T.; Burlingame, A.L.; Bechstedt, S.; et al. The Golgi Outpost Protein TPPP Nucleates Microtubules and Is Critical for Myelination. Cell 2019, 179, 132–146.e14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Target | Effect | Reference |
|---|---|---|---|
| PDpep1.3 | SYN | reduced SYN accumulation in C. elegans and rat models | [68] |
| Kisspeptin-10 1 | SYN | mitigated SYN-induced toxicity in SH-SY5Y cells | [69] |
| HER-096 2 | reduced SYN aggregation and modulated the unfolded protein response pathway in vitro and in a mouse model | [70] |
| Compound | Design | Effects | Reference |
|---|---|---|---|
| Anle138b-centered PROTACs | Anle138b is a SYN binder and aggregation inhibitor; lenalidomide and thalidomide as E3 ligase Cereblon ligand | decreased SYN aggregation in neuronal cells | [82] |
| sery384-based PROTACS | sery 384 is a SYN aggregation inhibitor; common ligands of E3 ligases (Cereblon, von Hippel–Lindau, cIAP1) | induced degradation of SYN aggregates in a dose- and time-dependent manner in cells | [83,84] |
| PD163916 | an established active-site inhibitor of p38 MAPK, resulting in proteasome activation | decreased SYN levels in cells | [80] |
| peptide TAT-PBD-PTM | targeting peptide with a cell-penetrating domain (TAT), a SYN protein-binding domain (PBD), and a short strong proteasome-targeting motif (PTM) | decreased the SYN level through proteasome degradation in a dose- and time-dependent manner in cultured cells and primary neurons | [85] |
| ATC161, a Autophagy-Targeting Chimera | bind both SYN aggregates (via Anle138b) and p62/SQSTM1/Sequestosome-1, an autophagic receptor | induced selective degradation of SYN aggregates but not monomers in cells and a mouse model; improved muscle strength and locomotive activity in mouse | [86] |
| Compound | Target | Effects | Reference |
|---|---|---|---|
| Synucleozid | SNCA mRNA 5′ UTR at the iron-responsive element | decreased SYN level | [99] |
| quercetin | pri-miR-7/HuR interaction inhibitor | upregulated miR-7, decreased SYN level | [100] |
| miR-7 | 3′-untranslated region of SNCA mRNA | reduced SYN levels, protected neuronal cells, and protected against preformed fibrils in mice | [97] |
| GRK2-specific miRNA | G-protein-coupled receptor kinase 2 (GRK2) | reduction in high-molecular-weight SYN species and neuroprotection in a mouse model | [101] |
| shRNA-minicircles delivered by RVG-extracellular vesicles | SYN downregulation in mice | [102] | |
| miRNA-206 | TPPP | downregulation of TPPP in progenitor OLGs 1 | [17] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oláh, J.; Norris, V.; Lehotzky, A.; Ovádi, J. Perspective Strategies for Interventions in Parkinsonism: Remedying the Neglected Role of TPPP. Cells 2024, 13, 338. https://doi.org/10.3390/cells13040338
Oláh J, Norris V, Lehotzky A, Ovádi J. Perspective Strategies for Interventions in Parkinsonism: Remedying the Neglected Role of TPPP. Cells. 2024; 13(4):338. https://doi.org/10.3390/cells13040338
Chicago/Turabian StyleOláh, Judit, Vic Norris, Attila Lehotzky, and Judit Ovádi. 2024. "Perspective Strategies for Interventions in Parkinsonism: Remedying the Neglected Role of TPPP" Cells 13, no. 4: 338. https://doi.org/10.3390/cells13040338
APA StyleOláh, J., Norris, V., Lehotzky, A., & Ovádi, J. (2024). Perspective Strategies for Interventions in Parkinsonism: Remedying the Neglected Role of TPPP. Cells, 13(4), 338. https://doi.org/10.3390/cells13040338