
Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Immunofluorescence Microscopy
2.3. Glutathione–S-Transferase (GST) Pull-Down
2.4. Quantitative Polymerase Chain Reaction (qPCR) and Western Blot Analysis
2.5. Time-Lapse Video Microscopy and Migration Analysis
2.6. Interference Reflection Microscopy (IRM)
2.7. Statistical Analysis
3. Results
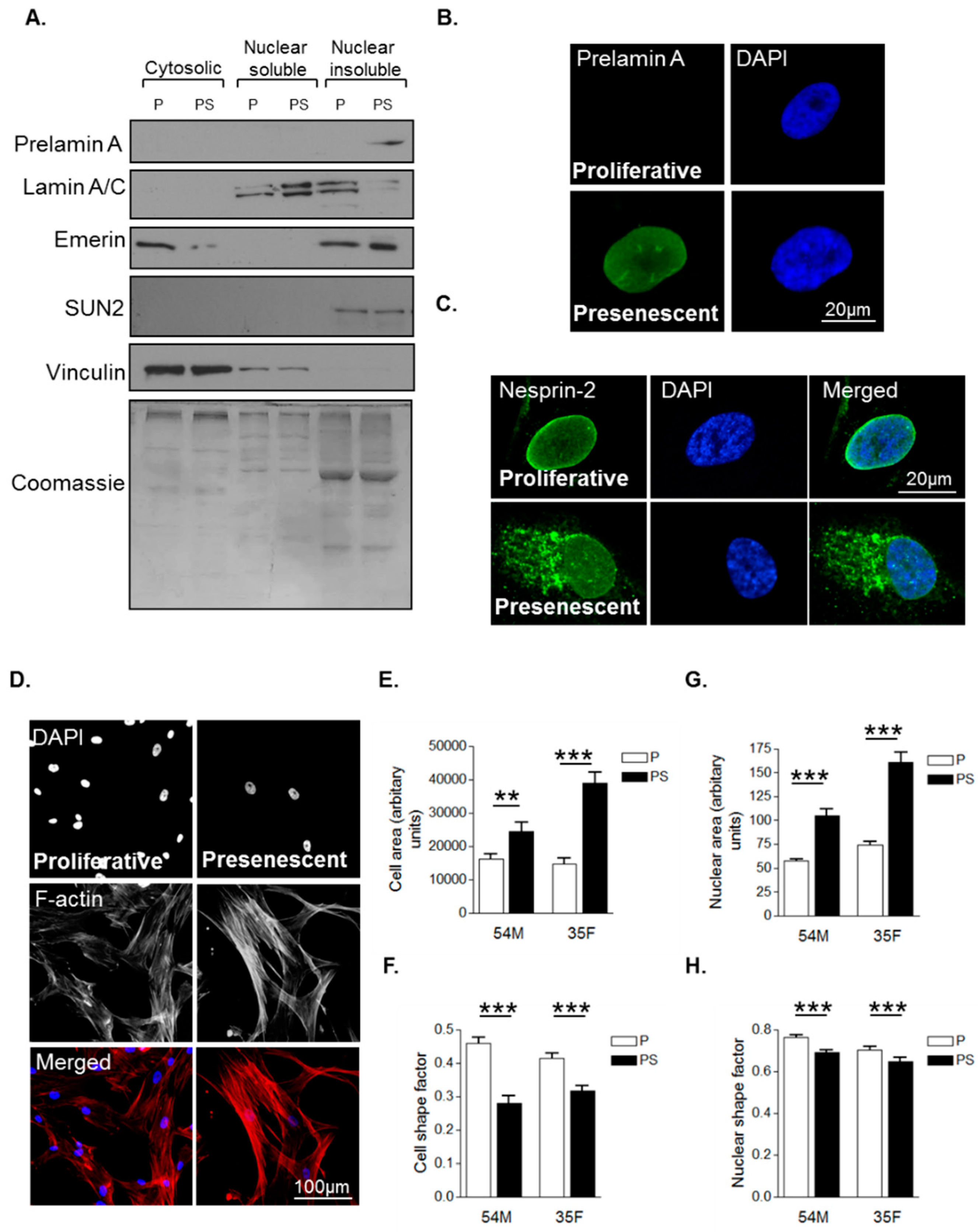
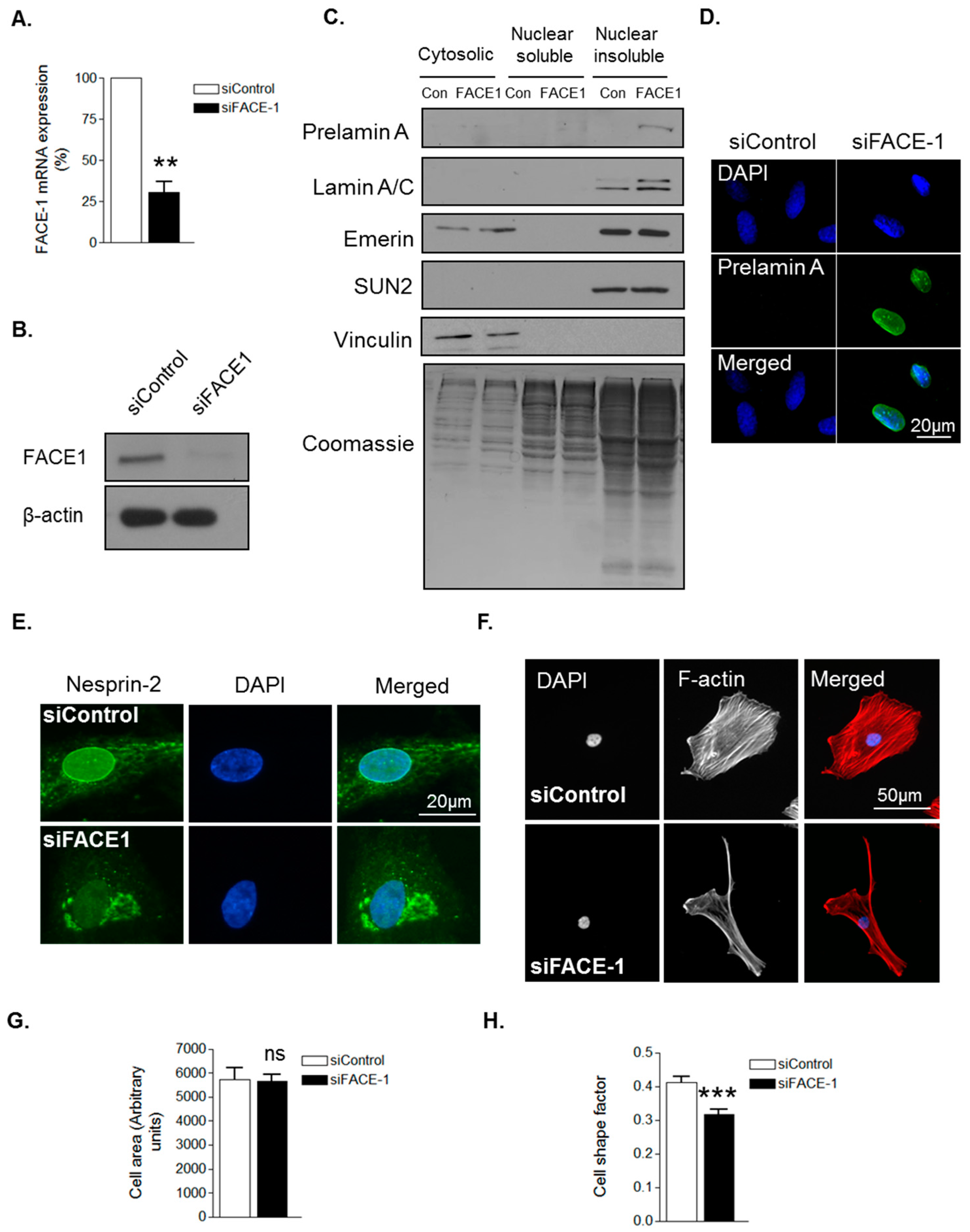
3.1. Prelamin A Accumulation Drives Aged-Related Morphological Changes
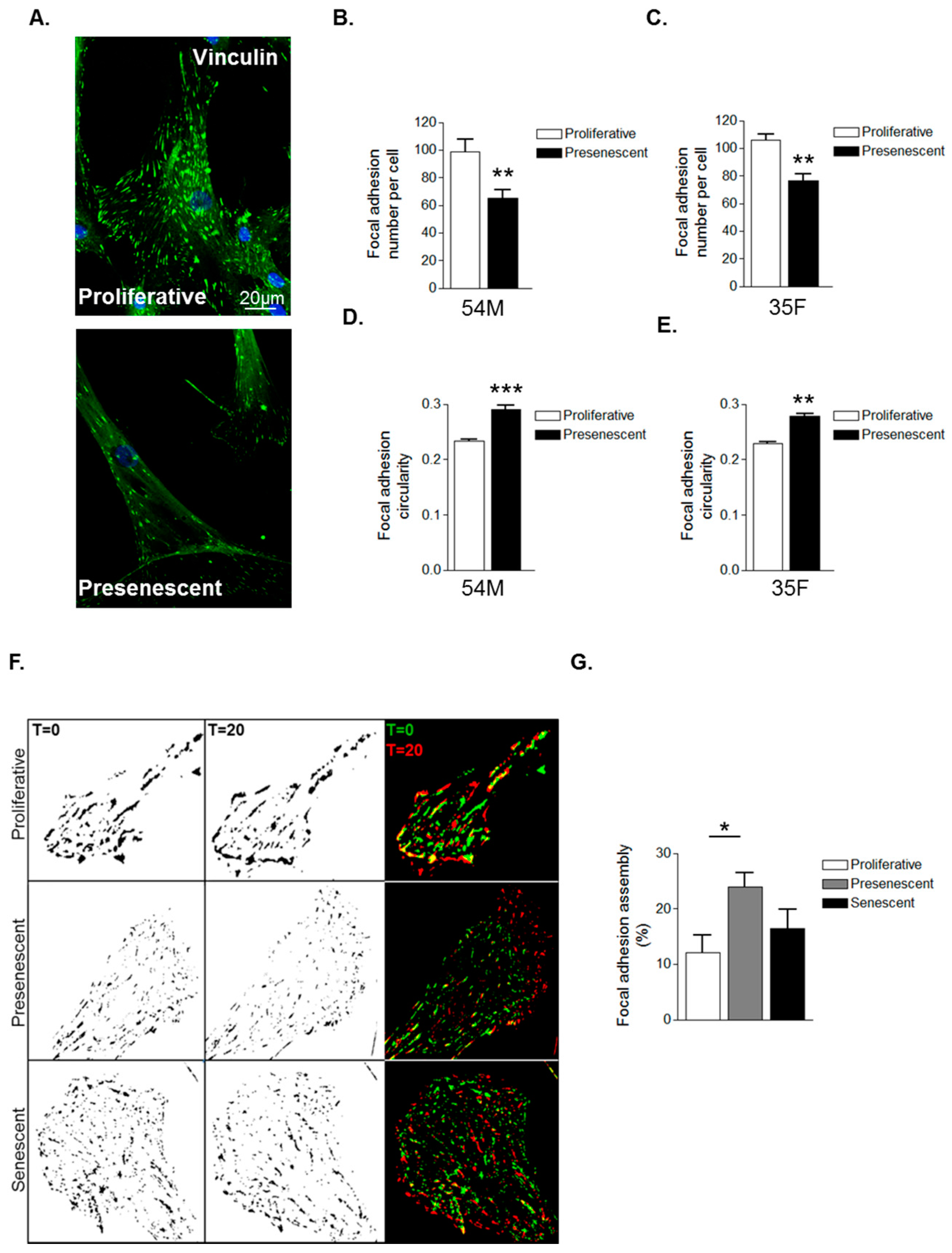
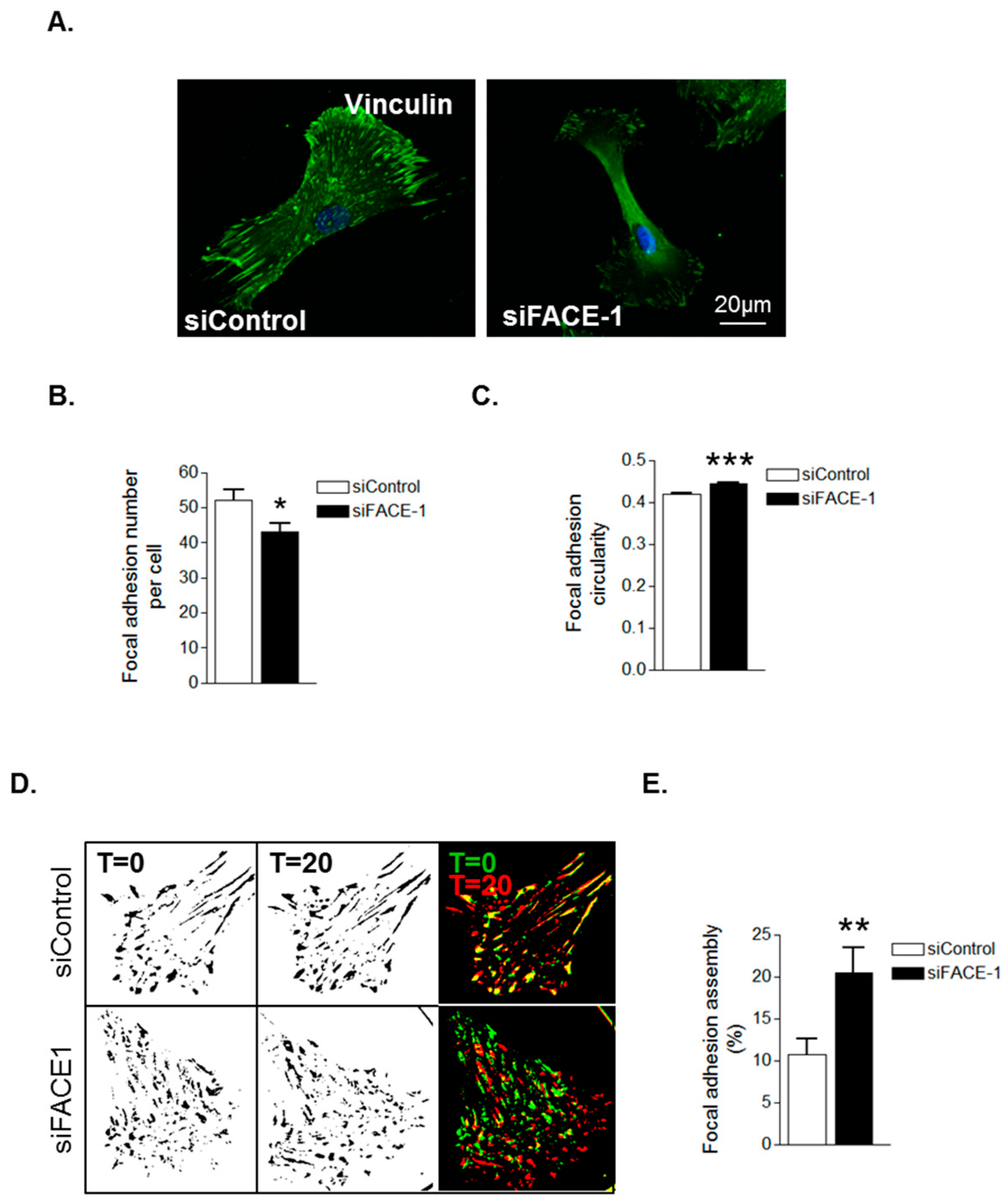
3.2. Prelamin A Accumulation Promotes Focal Adhesion Reorganisation
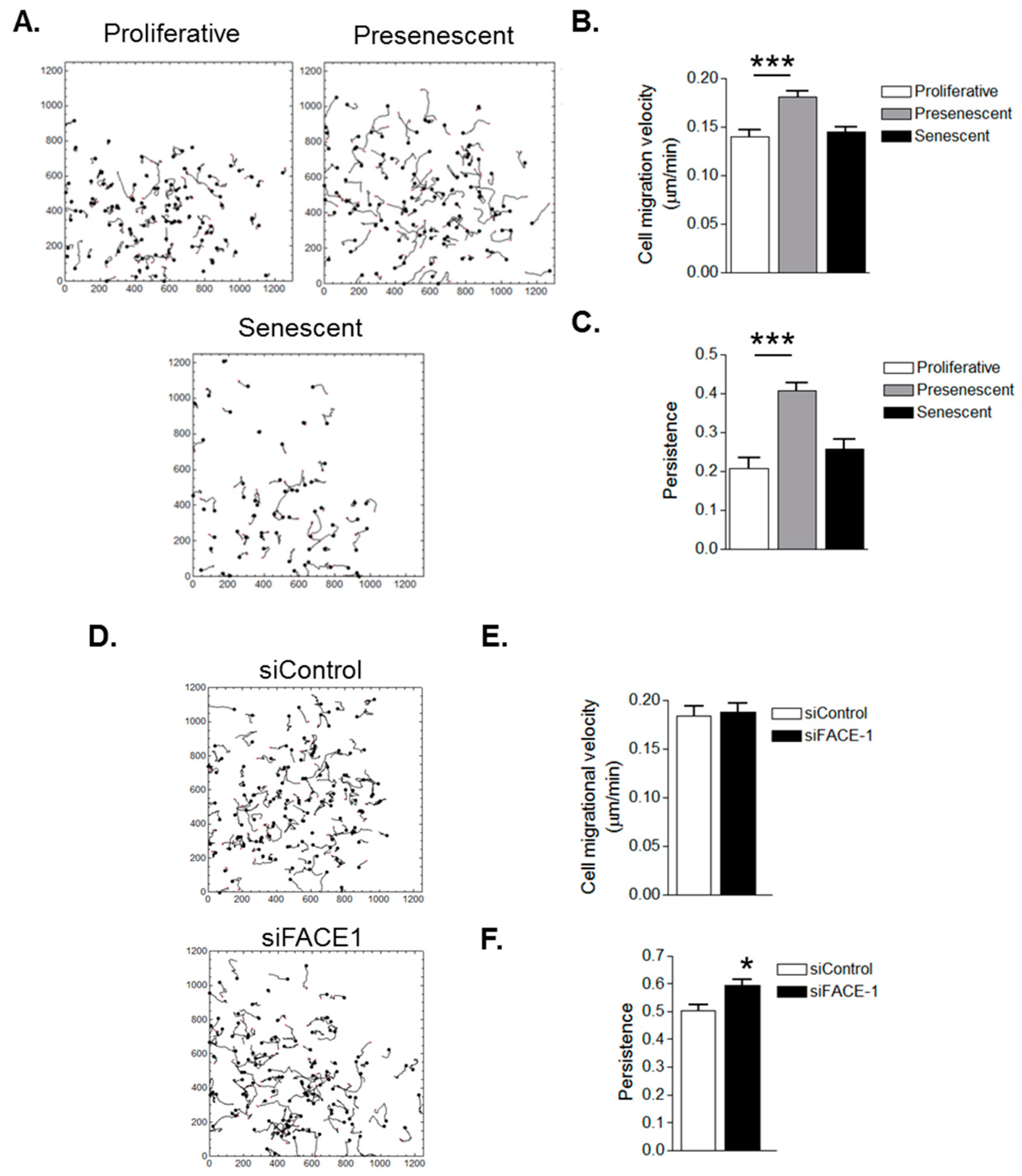
3.3. VSMC Ageing and Prelamin A Accumulation Stimulate Migrational Persistence
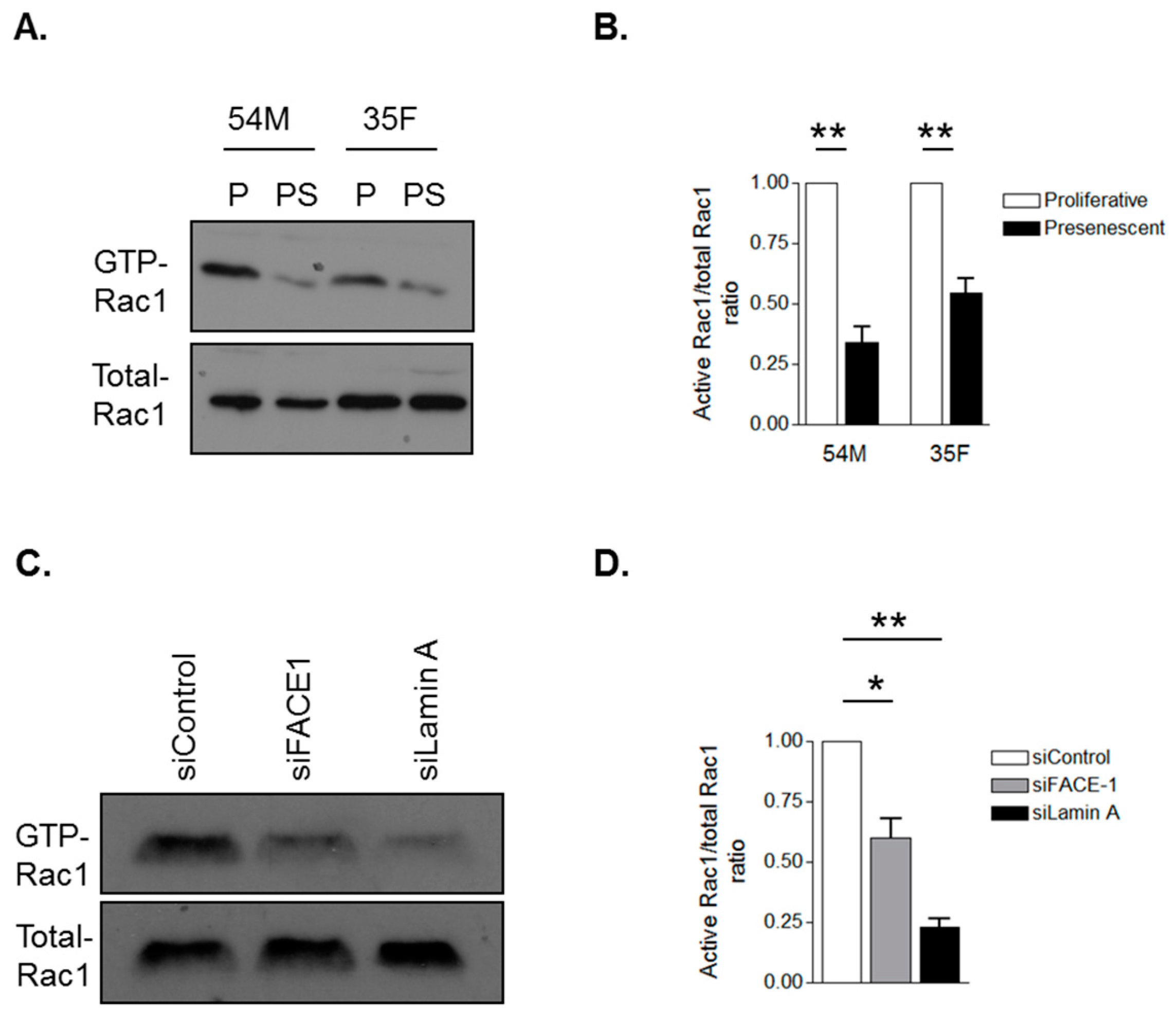
3.4. Prelamin A Accumulation Attenuates Rac1 Activity during VSMC Ageing
4. Discussion
Changes in Migrational Persistence during VSMC Ageing
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wissler, R.W.; Robert, L. Aging and cardiovascular disease: A summary of the Eighth Munster International Arteriosclerosis Symposium. Circulation 1996, 93, 1608–1612. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part III: Cellular and molecular clues to heart and arterial aging. Circulation 2003, 107, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Tahir, H.; Niculescu, I.; Bona-Casas, C.; Merks, R.M.; Hoekstra, A.G. An in silico study on the role of smooth muscle cell migration in neointimal formation after coronary stenting. J. R. Soc. Interface 2015, 12, 20150358. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Nagel, D.J.; Zhou, Q.; Cygnar, K.D.; Zhao, H.; Li, F.; Pi, X.; Knight, P.A.; Yan, C. Role of cAMP-phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ. Res. 2015, 116, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Chen, S.Y. Smooth Muscle Cell Differentiation: Model Systems, Regulatory Mechanisms, and Vascular Diseases. J. Cell Physiol. 2015, 231, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Fabbiano, S.; Menacho-Marquez, M.; Sevilla, M.A.; Albarran-Juarez, J.; Zheng, Y.; Offermanns, S.; Montero, M.J.; Bustelo, X.R. Genetic dissection of the vav2-rac1 signaling axis in vascular smooth muscle cells. Mol. Cell Biol. 2014, 34, 4404–4419. [Google Scholar] [CrossRef] [PubMed]
- Pankov, R.; Endo, Y.; Even-Ram, S.; Araki, M.; Clark, K.; Cukierman, E.; Matsumoto, K.; Yamada, K.M. A Rac switch regulates random versus directionally persistent cell migration. J. Cell Biol. 2005, 170, 793–802. [Google Scholar] [CrossRef] [PubMed]
- Chambliss, A.B.; Khatau, S.B.; Erdenberger, N.; Robinson, D.K.; Hodzic, D.; Longmore, G.D.; Wirtz, D. The LINC-anchored actin cap connects the extracellular milieu to the nucleus for ultrafast mechanotransduction. Sci. Rep. 2013, 3, 1087. [Google Scholar] [CrossRef] [PubMed]
- Crisp, M.; Liu, Q.; Roux, K.; Rattner, J.B.; Shanahan, C.; Burke, B.; Stahl, P.D.; Hodzic, D. Coupling of the nucleus and cytoplasm: Role of the LINC complex. J. Cell Biol. 2006, 172, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Osborne, L.D.; Van Landeghem, L.; Sharek, L.; Superfine, R.; Garcia-Mata, R.; Burridge, K. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 2014, 16, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, M.L.; Jaalouk, D.E.; Shanahan, C.M.; Burke, B.; Roux, K.J.; Lammerding, J. The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J. Biol. Chem. 2011, 286, 26743–26753. [Google Scholar] [CrossRef] [PubMed]
- Young, S.G.; Fong, L.G.; Michaelis, S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria—New evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid. Res. 2005, 46, 2531–2558. [Google Scholar] [CrossRef] [PubMed]
- Rusinol, A.E.; Sinensky, M.S. Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors. J. Cell Sci. 2006, 119, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, D.P.; Kuszczak, D.; Rusinol, A.E.; Thewke, D.P.; Hrycyna, C.A.; Michaelis, S.; Sinensky, M.S. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem. J. 2005, 387, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Kudlow, B.A.; Kennedy, B.K.; Monnat, R.J., Jr. Werner and Hutchinson-Gilford progeria syndromes: Mechanistic basis of human progeroid diseases. Nat. Rev. Mol. Cell Biol. 2007, 8, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Shumaker, D.K.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Gordon, L.B.; Gruenbaum, Y.; Khuon, S.; Mendez, M.; Varga, R.; et al. Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 8963–8968. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat. Med. 2005, 11, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Risques, R.A.; Martin, G.M.; Rabinovitch, P.S.; Oshima, J. Accelerated telomere shortening and replicative senescence in human fibroblasts overexpressing mutant and wild-type lamin A. Exp. Cell Res. 2008, 314, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Kutscheidt, S.; Zhu, R.; Antoku, S.; Luxton, G.W.; Stagljar, I.; Fackler, O.T.; Gundersen, G.G. FHOD1 interaction with nesprin-2G mediates TAN line formation and nuclear movement. Nat. Cell Biol. 2014, 16, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Dechat, T.; Pfleghaar, K.; Sengupta, K.; Shimi, T.; Shumaker, D.K.; Solimando, L.; Goldman, R.D. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008, 22, 832–853. [Google Scholar] [CrossRef] [PubMed]
- Merideth, M.A.; Gordon, L.B.; Clauss, S.; Sachdev, V.; Smith, A.C.; Perry, M.B.; Brewer, C.C.; Zalewski, C.; Kim, H.J.; Solomon, B.; et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Bergo, M.O.; Gavino, B.; Ross, J.; Schmidt, W.K.; Hong, C.; Kendall, L.V.; Mohr, A.; Meta, M.; Genant, H.; Jiang, Y.; et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl. Acad. Sci. USA 2002, 99, 13049–13054. [Google Scholar] [CrossRef] [PubMed]
- Ragnauth, C.D.; Warren, D.T.; Liu, Y.; McNair, R.; Tajsic, T.; Figg, N.; Shroff, R.; Skepper, J.; Shanahan, C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation 2010, 121, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Warren, D.T.; Tajsic, T.; Porter, L.J.; Minaisah, R.M.; Cobb, A.; Jacob, A.; Rajgor, D.; Zhang, Q.P.; Shanahan, C.M. Nesprin-2-dependent ERK1/2 compartmentalisation regulates the DNA damage response in vascular smooth muscle cell ageing. Cell Death Differ. 2015, 22, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, L.M.; Gomez, L.A.; Dias, J.; Ziebarth, N.M.; Howard, G.A.; Schiller, P.C. Progerin expression disrupts critical adult stem cell functions involved in tissue repair. Aging (Albany NY) 2014, 6, 1049–1063. [Google Scholar] [CrossRef] [PubMed]
- Booth-Gauthier, E.A.; Du, V.; Ghibaudo, M.; Rape, A.D.; Dahl, K.N.; Ladoux, B. Hutchinson-Gilford progeria syndrome alters nuclear shape and reduces cell motility in three dimensional model substrates. Integr. Biol. 2013, 5, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Shanahan, C. Human vascular smooth muscle cell culture. Methods Mol. Biol. 2012, 806, 251–263. [Google Scholar] [PubMed]
- Zhang, Q.; Minaisah, R.M.; Ferraro, E.; Li, C.; Porter, L.J.; Zhou, C.; Gao, F.; Zhang, J.; Rajgor, D.; Autore, F.; et al. N-terminal nesprin-2 variants regulate beta-catenin signalling. Exp. Cell Res. 2016, 345, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.R.; Calle, Y.; Sutton, D.H.; Critchley, D.R.; Jones, G.E.; Dunn, G.A. Quantifying cell-matrix adhesion dynamics in living cells using interference reflection microscopy. J. Microsc. 2008, 232, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Mayanagi, T.; Sobue, K. Diversification of caldesmon-linked actin cytoskeleton in cell motility. Cell Adhes. Migr. 2011, 5, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Haque, F.; Lloyd, D.J.; Smallwood, D.T.; Dent, C.L.; Shanahan, C.M.; Fry, A.M.; Trembath, R.C.; Shackleton, S. SUN1 interacts with nuclear lamin A and cytoplasmic nesprins to provide a physical connection between the nuclear lamina and the cytoskeleton. Mol. Cell Biol. 2006, 26, 3738–3751. [Google Scholar] [CrossRef] [PubMed]
- Khatau, S.B.; Kusuma, S.; Hanjaya-Putra, D.; Mali, P.; Cheng, L.; Lee, J.S.; Gerecht, S.; Wirtz, D. The differential formation of the LINC-mediated perinuclear actin cap in pluripotent and somatic cells. PLoS ONE 2012, 7, e36689. [Google Scholar] [CrossRef] [PubMed]
- Lammerding, J.; Schulze, P.C.; Takahashi, T.; Kozlov, S.; Sullivan, T.; Kamm, R.D.; Stewart, C.L.; Lee, R.T. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Investig. 2004, 113, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Swift, J.; Ivanovska, I.L.; Buxboim, A.; Harada, T.; Dingal, P.C.; Pinter, J.; Pajerowski, J.D.; Spinler, K.R.; Shin, J.W.; Tewari, M.; et al. Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 2013, 341, 1240104. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, V.L.; Ji, J.Y.; Cummings, K.S.; Lee, R.T.; Lammerding, J. Increased mechanosensitivity and nuclear stiffness in Hutchinson-Gilford progeria cells: Effects of farnesyltransferase inhibitors. Aging Cell 2008, 7, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Rocnik, E.F.; Chan, B.M.; Pickering, J.G. Evidence for a role of collagen synthesis in arterial smooth muscle cell migration. J. Clin. Investig. 1998, 101, 1889–1898. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Chow, L.H.; Pickering, J.G. Cell surface-bound collagenase-1 and focal substrate degradation stimulate the rear release of motile vascular smooth muscle cells. J. Biol. Chem. 2000, 275, 35384–35392. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.C.; Gotlieb, A.I. Towards understanding acute destabilization of vulnerable atherosclerotic plaques. Cardiovasc. Pathol. 2003, 12, 237–248. [Google Scholar] [CrossRef]
- Naghavi, M.; Libby, P.; Falk, E.; Casscells, S.W.; Litovsky, S.; Rumberger, J.; Badimon, J.J.; Stefanadis, C.; Moreno, P.; Pasterkamp, G.; et al. From vulnerable plaque to vulnerable patient: A call for new definitions and risk assessment strategies: Part II. Circulation 2003, 108, 1772–1778. [Google Scholar] [CrossRef] [PubMed]
- Izzo, J.L., Jr.; Shykoff, B.E. Arterial stiffness: Clinical relevance, measurement, and treatment. Rev. Cardiovasc. Med. 2001, 2, 29–40. [Google Scholar] [PubMed]
- Orlandi, A.; Marcellini, M.; Spagnoli, L.G. Aging influences development and progression of early aortic atherosclerotic lesions in cholesterol-fed rabbits. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Louis, S.F.; Zahradka, P. Vascular smooth muscle cell motility: From migration to invasion. Exp. Clin. Cardiol. 2010, 15, e75–e85. [Google Scholar] [PubMed]






© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, L.J.; Holt, M.R.; Soong, D.; Shanahan, C.M.; Warren, D.T. Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells. Cells 2016, 5, 41. https://doi.org/10.3390/cells5040041
Porter LJ, Holt MR, Soong D, Shanahan CM, Warren DT. Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells. Cells. 2016; 5(4):41. https://doi.org/10.3390/cells5040041
Chicago/Turabian StylePorter, Lauren J., Mark R. Holt, Daniel Soong, Catherine M. Shanahan, and Derek T. Warren. 2016. "Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells" Cells 5, no. 4: 41. https://doi.org/10.3390/cells5040041
APA StylePorter, L. J., Holt, M. R., Soong, D., Shanahan, C. M., & Warren, D. T. (2016). Prelamin A Accumulation Attenuates Rac1 Activity and Increases the Intrinsic Migrational Persistence of Aged Vascular Smooth Muscle Cells. Cells, 5(4), 41. https://doi.org/10.3390/cells5040041