
May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells

Abstract
:1. Introduction
2. Gene Targeting by Homologous Recombination (HR)
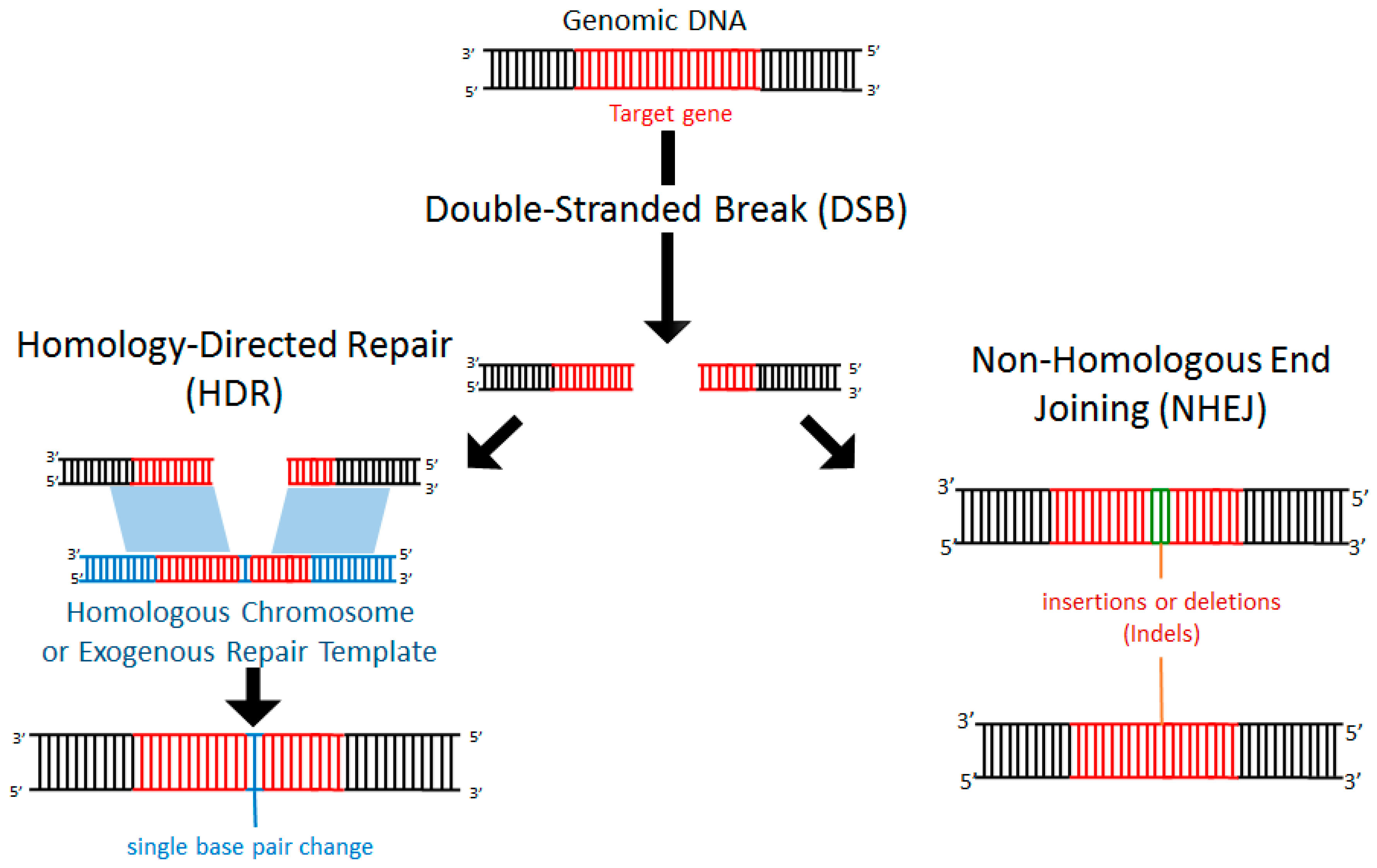
3. Endogenous Repair of Double-Stranded DNA Breaks (DSB)
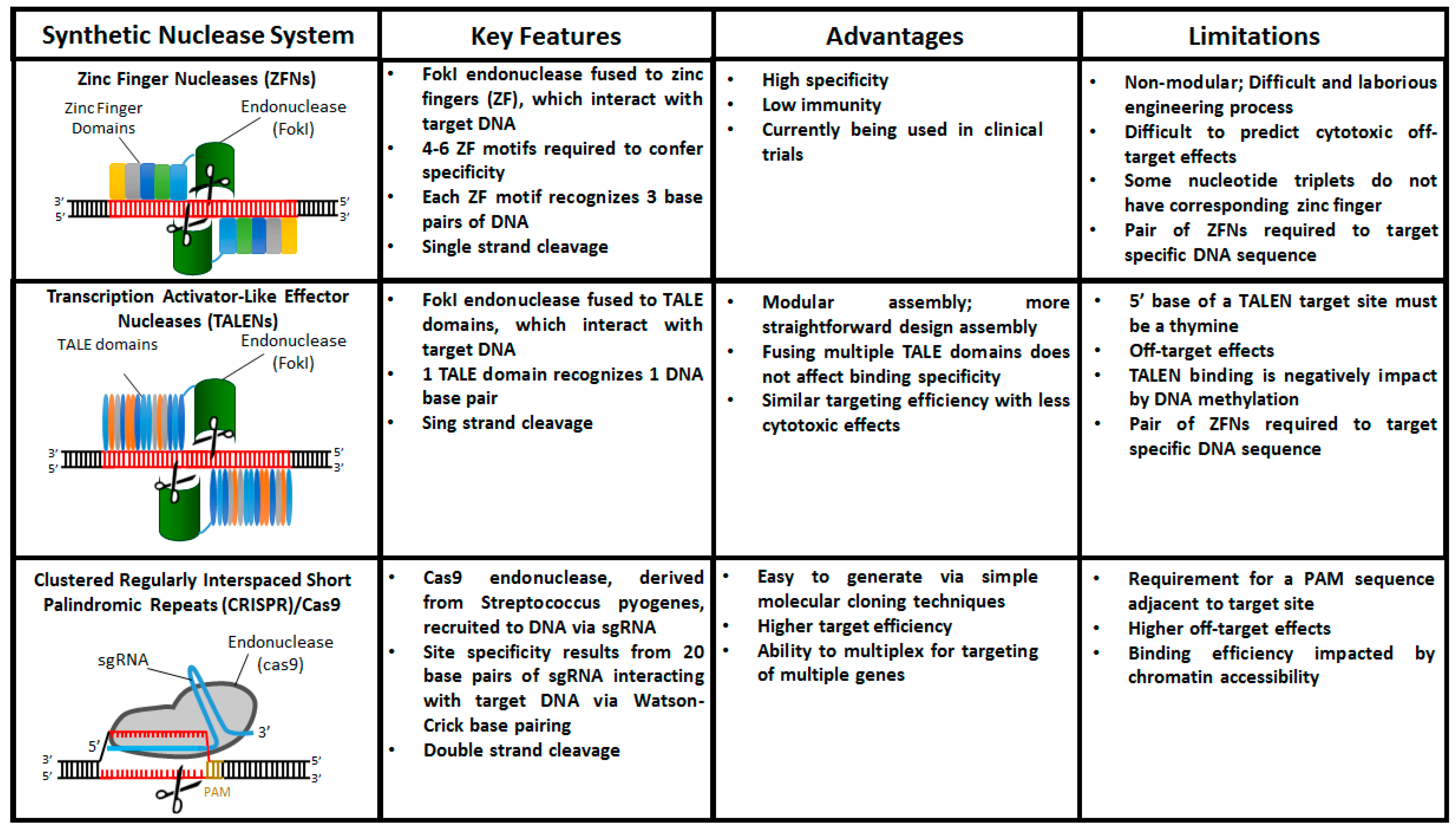
4. The Age of Designer Nucleases: ZFNs, TALENs, and CRISPR/Cas9
4.1. Zinc Finger Nucleases (ZFNs)
4.2. Transcription Activator-Like Effector Nucleases (TALENs)
4.3. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9
4.4. Practical Considerations for Synthetic Nuclease Mediated HR
5. Applications of Gene Editing Technologies with hiPSCs
5.1. Generation of Targeted Reporter Lines
5.2. Using Designer Nucleases to Generate hiPSC-Based Disease Models
5.2.1. ZFN
5.2.2. TALENs
5.2.3. CRISPR/Cas9
5.3. Development of Gene Edited hiPSCs for Cell-Based Therapies
6. Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, O.; Habib, G.; Choi, H.W.; Hong, K.S.; Do, J.T.; Moon, S.H.; Chung, H.M. An improved method for the derivation of high quality iPSCs in the absence of c-myc. Exp. Cell Res. 2013, 319, 3190–3200. [Google Scholar] [CrossRef] [PubMed]
- Su, R.J.; Baylink, D.J.; Neises, A.; Kiroyan, J.B.; Meng, X.; Payne, K.J.; Tschudy-Seney, B.; Duan, Y.; Appleby, N.; Kearns-Jonker, M.; et al. Efficient generation of integration-free ips cells from human adult peripheral blood using bcl-xl together with yamanaka factors. PLoS ONE 2013, 8, e64496. [Google Scholar] [CrossRef] [PubMed]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Wu, S.; Joo, J.Y.; Zhu, S.; Han, D.W.; Lin, T.; Trauger, S.; Bien, G.; Yao, S.; Zhu, Y.; et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell 2009, 4, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Li, Y.; Zhang, X.; Liu, C.; Guan, J.; Li, H.; Zhao, T.; Ye, J.; Yang, W.; Liu, K.; et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science 2013, 341, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Maehr, R.; Guo, W.; Eijkelenboom, A.; Snitow, M.; Chen, A.E.; Melton, D.A. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat. Biotechnol. 2008, 26, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.; Ambasudhan, R.; Yuan, X.; Li, W.; Hilcove, S.; Abujarour, R.; Lin, X.; Hahm, H.S.; Hao, E.; Hayek, A.; et al. A chemical platform for improved induction of human iPSCs. Nat. Methods 2009, 6, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Zhu, X.; Liao, B.; Benda, C.; Zhuang, Q.; Pei, D.; Qin, B.; Esteban, M.A. Micrornas in somatic cell reprogramming. Curr. Opin. Cell Biol. 2013, 25, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Srivastava, A.; Srivastava, P.; Dhuriya, Y.K.; Pandey, A.; Kumar, D.; Rajpurohit, C.S. Advances in stem cell research- a ray of hope in better diagnosis and prognosis in neurodegenerative diseases. Front. Mol. Biosci. 2016, 3, 72. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Jiao, B.; Zhou, M.; Zhou, T.; Shen, L. Modeling Alzheimer's disease with induced pluripotent stem cells: Current challenges and future concerns. Stem Cells Int. 2016, 2016, 7828049. [Google Scholar] [CrossRef] [PubMed]
- Preza, E.; Hardy, J.; Warner, T.; Wray, S. Review: Induced pluripotent stem cell models of frontotemporal dementia. Neuropathol. Appl. Neurobiol. 2016, 42, 497–520. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, S.; He, X.B.; Cheng, C.; Le, W. Induced pluripotent stem cells in Alzheimer's Disease: Applications for disease modeling and cell-replacement therapy. Mol. Neurodegener. 2016, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Mungenast, A.E.; Siegert, S.; Tsai, L.H. Modeling Alzheimer’s disease with human induced pluripotent stem (iPS) cells. Mol. Cell. Neurosci. 2016, 73, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Young-Pearse, T.L.; Morrow, E.M. Modeling developmental neuropsychiatric disorders with iPSC technology: Challenges and opportunities. Curr. Opin. Neurobiol. 2016, 36, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Matsa, E.; Ahrens, J.H.; Wu, J.C. Human induced pluripotent stem cells as a platform for personalized and precision cardiovascular medicine. Physiol. Rev. 2016, 96, 1093–1126. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yuasa, S.; Node, K.; Fukuda, K. Cardiovascular disease modeling using patient-specific induced pluripotent stem cells. Int. J. Mol. Sci. 2015, 16, 18894–18922. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.S. Modeling kidney disease with ips cells. Biomark. Insights 2015, 10, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Mou, H.; Brazauskas, K.; Rajagopal, J. Personalized medicine for cystic fibrosis: Establishing human model systems. Pediatr. Pulmonol. 2015, 50 (Suppl. S40), S14–S23. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Wang, D. Clinical potentials of human pluripotent stem cells in lung diseases. Clin. Transl. Med. 2014, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Golas, M.M.; Sander, B. Use of human stem cells in huntington disease modeling and translational research. Exp. Neurol. 2016, 278, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Kawser Hossain, M.; Abdal Dayem, A.; Han, J.; Kumar Saha, S.; Yang, G.M.; Choi, H.Y.; Cho, S.G. Recent advances in disease modeling and drug discovery for diabetes mellitus using induced pluripotent stem cells. Int. J. Mol. Sci. 2016, 17, 256. [Google Scholar] [CrossRef] [PubMed]
- Balboa, D.; Otonkoski, T. Human pluripotent stem cell based islet models for diabetes research. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Goto, Y. Concise review: Heteroplasmic mitochondrial DNA mutations and mitochondrial diseases: Toward iPSC-based disease modeling, drug discovery, and regenerative therapeutics. Stem Cells 2016, 34, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.C.; Chen, C.T.; Wei, Y.H. Mitochondrial resetting and metabolic reprogramming in induced pluripotent stem cells and mitochondrial disease modeling. Biochim. Biophys. Acta. 2016, 1860, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Kalsan, M.; Kumar, N.; Saini, A.; Chandra, R. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front. Cell Dev. Biol. 2015, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- RIKEN; Japan. A Study of transplantation of autologous induced pluripotent stem cell (iPSC) derived retinal pigment epithelium (RPE) cell sheet in subjects with exudative age related macular degeneration. umin.ac.jp; Japan, 2013–16 December 2016. Available online: https://upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000013279. UMIN ID : UMIN000011929 (accessed on 2 March 2017).
- Hall, B.; Limaye, A.; Kulkarni, A.B. Overview: Generation of gene knockout mice. Curr. Protoc. Cell Biol. 2009, 19, 11–17. [Google Scholar]
- Sakamoto, K.; Gurumurthy, C.B.; Wagner, K.U. Generation of conditional knockout mice. Methods Mol. Biol. 2014, 1194, 21–35. [Google Scholar] [PubMed]
- Tong, C.; Huang, G.; Ashton, C.; Li, P.; Ying, Q.L. Generating gene knockout rats by homologous recombination in embryonic stem cells. Nat. Protoc. 2011, 6, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Te Riele, H.; Maandag, E.R.; Berns, A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc. Natl. Acad. Sci. USA 1992, 89, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M. Gene targeting in human cells without isogenic DNA. Science 1999, 283, 9. [Google Scholar] [CrossRef]
- Urbach, A.; Schuldiner, M.; Benvenisty, N. Modeling for lesch-nyhan disease by gene targeting in human embryonic stem cells. Stem Cells 2004, 22, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Dutriaux, A. Gene targeting and somatic cell genetics--a rebirth or a coming of age? Trends Genet. 1999, 15, 88–90. [Google Scholar] [CrossRef]
- Ruby, K.M.; Zheng, B. Gene targeting in a hues line of human embryonic stem cells via electroporation. Stem Cells 2009, 27, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Zwaka, T.P.; Thomson, J.A. Homologous recombination in human embryonic stem cells. Nat. Biotechnol. 2003, 21, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, A.I.; Christodoulou, I.; Pells, S.C.; McWhir, J.; Thomson, A.J. Sequential genetic modification of the hprt locus in human escs combining gene targeting and recombinase-mediated cassette exchange. Cloning Stem Cells 2008, 10, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Irion, S.; Luche, H.; Gadue, P.; Fehling, H.J.; Kennedy, M.; Keller, G. Identification and targeting of the rosa26 locus in human embryonic stem cells. Nat. Biotechnol. 2007, 25, 1477–1482. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Ueno, M.; Kamiya, D.; Nishiyama, A.; Matsumura, M.; Wataya, T.; Takahashi, J.B.; Nishikawa, S.; Nishikawa, S.; Muguruma, K.; et al. A rock inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol. 2007, 25, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Merkert, S.; Martin, U. Site-specific genome engineering in human pluripotent stem cells. Int. J. Mol. Sci. 2016, 17, 1000. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.H.; Izpisua Belmonte, J.C. A cut above the rest: Targeted genome editing technologies in human pluripotent stem cells. J. Biol. Chem. 2014, 289, 4594–4599. [Google Scholar] [CrossRef] [PubMed]
- Banuelos, C.A.; Banath, J.P.; MacPhail, S.H.; Zhao, J.; Eaves, C.A.; O’Connor, M.D.; Lansdorp, P.M.; Olive, P.L. Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair (Amst.) 2008, 7, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- De Los Angeles, A.; Loh, Y.H.; Tesar, P.J.; Daley, G.Q. Accessing naive human pluripotency. Curr. Opin. Genet. Dev. 2012, 22, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Gafni, O.; Weinberger, L.; Mansour, A.A.; Manor, Y.S.; Chomsky, E.; Ben-Yosef, D.; Kalma, Y.; Viukov, S.; Maza, I.; Zviran, A.; et al. Derivation of novel human ground state naive pluripotent stem cells. Nature 2013, 504, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Argueso, J.L.; Westmoreland, J.; Mieczkowski, P.A.; Gawel, M.; Petes, T.D.; Resnick, M.A. Double-strand breaks associated with repetitive DNA can reshape the genome. Proc. Natl. Acad. Sci. USA 2008, 105, 11845–11850. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Seah, Y.F.; El Farran, C.A.; Warrier, T.; Xu, J.; Loh, Y.H. Induced pluripotency and gene editing in disease modelling: Perspectives and challenges. Int. J. Mol. Sci. 2015, 16, 28614–28634. [Google Scholar] [CrossRef] [PubMed]
- Pardo, B.; Gomez-Gonzalez, B.; Aguilera, A. DNA repair in mammalian cells: DNA double-strand break repair: How to fix a broken relationship. Cell. Mol. Life Sci. 2009, 66, 1039–1056. [Google Scholar] [CrossRef] [PubMed]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol. Cell. Biol. 1994, 14, 8096–8106. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. USA 1994, 91, 6064–6068. [Google Scholar] [CrossRef] [PubMed]
- Smih, F.; Rouet, P.; Romanienko, P.J.; Jasin, M. Double-strand breaks at the target locus stimulate gene targeting in embryonic stem cells. Nucleic Acids Res. 1995, 23, 5012–5019. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome engineering with zinc-finger nucleases. Genetics 2011, 188, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Segal, D.J.; Crotty, J.W.; Bhakta, M.S.; Barbas, C.F., 3rd; Horton, N.C. Structure of aart, a designed six-finger zinc finger peptide, bound to DNA. J. Mol. Biol. 2006, 363, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Smith, J.; Kandavelou, K.; Berg, J.M.; Chandrasegaran, S. Binding of two zinc finger nuclease monomers to two specific sites is required for effective double-strand DNA cleavage. Biochem. Biophys. Res. Commun. 2005, 334, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Porteus, M.H.; Baltimore, D. Chimeric nucleases stimulate gene targeting in human cells. Science 2003, 300, 763. [Google Scholar] [CrossRef] [PubMed]
- Hofer, U.; Henley, J.E.; Exline, C.M.; Mulhern, O.; Lopez, E.; Cannon, P.M. Pre-clinical modeling of CCR5 knockout in human hematopoietic stem cells by zinc finger nucleases using humanized mice. J. Infect. Dis 2013, 208 (Suppl. S2), S160–164. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of hiv-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with hiv. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Isalan, M. Zinc-finger nucleases: How to play two good hands. Nat. Methods 2011, 9, 32–34. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, R.; Lombardo, A.; Arens, A.; Miller, J.C.; Genovese, P.; Kaeppel, C.; Nowrouzi, A.; Bartholomae, C.C.; Wang, J.; Friedman, G.; et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat. Biotechnol. 2011, 29, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Muller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Thibodeau-Beganny, S.; Osiak, A.; Wright, D.A.; Anthony, R.M.; Eichtinger, M.; Jiang, T.; Foley, J.E.; Winfrey, R.J.; Townsend, J.A.; et al. Rapid "open-source" engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol. Cell. 2008, 31, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Bonas, U. Xanthomonas AvrBs3 family-type III effectors: Discovery and function. Annu. Rev. Phytopathol. 2010, 48, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of tal-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A tale nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Cong, L.; Lodato, S.; Kosuri, S.; Church, G.M.; Arlotta, P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat. Biotechnol. 2011, 29, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, A.W.; Rios, X.; Chari, R.; Yang, L.; Zhang, F.; Mali, P.; Church, G.M. Iterative capped assembly: Rapid and scalable synthesis of repeat-module DNA such as TAL effectors from individual monomers. Nucleic Acids Res. 2012, 40, e117. [Google Scholar] [CrossRef] [PubMed]
- Schmid-Burgk, J.L.; Schmidt, T.; Kaiser, V.; Honing, K.; Hornung, V. A ligation-independent cloning technique for high-throughput assembly of transcription activator-like effector genes. Nat. Biotechnol. 2013, 31, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kweon, J.; Kim, A.; Chon, J.K.; Yoo, J.Y.; Kim, H.J.; Kim, S.; Lee, C.; Jeong, E.; Chung, E.; et al. A library of tal effector nucleases spanning the human genome. Nat. Biotechnol. 2013, 31, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel tale nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef] [PubMed]
- Grau, J.; Boch, J.; Posch, S. TALENoffer: Genome-wide TALEN off-target prediction. Bioinformatics 2013, 29, 2931–2932. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Bultmann, S.; Morbitzer, R.; Schmidt, C.S.; Thanisch, K.; Spada, F.; Elsaesser, J.; Lahaye, T.; Leonhardt, H. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer tales and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012, 40, 5368–5377. [Google Scholar] [CrossRef] [PubMed]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; D'Souza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Marraffini, L.A.; Sontheimer, E.J. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat. Rev. Genet. 2010, 11, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. Elife. 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Musunuru, K. Genome engineering tools for building cellular models of disease. FEBS J. 2016, 283, 3222–3231. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Certo, M.T.; Ryu, B.Y.; Annis, J.E.; Garibov, M.; Jarjour, J.; Rawlings, D.J.; Scharenberg, A.M. Tracking genome engineering outcome at individual DNA breakpoints. Nat. Methods 2011, 8, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to foki nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Howden, S.E.; McColl, B.; Glaser, A.; Vadolas, J.; Petrou, S.; Little, M.H.; Elefanty, A.G.; Stanley, E.G. A Cas9 variant for efficient generation of indel-free knockin or gene-corrected human pluripotent stem cells. Stem Cell Rep. 2016, 7, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crrna array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Bernitz, J.M.; Lee, D.F.; Lemischka, I.R. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev. 2014, 23, 2673–2686. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Rivera-Perez, J.; Bradley, A. The length of homology required for gene targeting in embryonic stem cells. Mol. Cell. Biol. 1991, 11, 5586–5591. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Capecchi, M.R. Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol. Cell. Biol. 1992, 12, 3365–3371. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Chung, S.K.; Xu, Y. Modeling disease in human escs using an efficient BAC-based homologous recombination system. Cell Stem Cell 2010, 6, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A.; Court, D.L. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001, 2, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Bartel, M.A.; Schaffer, D.V. Enhanced gene targeting of adult and pluripotent stem cells using evolved adeno-associated virus. Methods Mol. Biol. 2014, 1114, 169–179. [Google Scholar] [PubMed]
- Khan, I.F.; Hirata, R.K.; Russell, D.W. AAV-mediated gene targeting methods for human cells. Nat. Protoc. 2011, 6, 482–501. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.F.; Hirata, R.K.; Wang, P.R.; Li, Y.; Kho, J.; Nelson, A.; Huo, Y.; Zavaljevski, M.; Ware, C.; Russell, D.W. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol. Ther. 2010, 18, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Brafman, D.A.; Moya, N.; Allen-Soltero, S.; Fellner, T.; Robinson, M.; McMillen, Z.L.; Gaasterland, T.; Willert, K. Analysis of SOX2-expressing cell populations derived from human pluripotent stem cells. Stem Cell Rep. 2013, 1, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Vasileva, A.; Jessberger, R. Precise hit: Adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005, 3, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Nolden, L.; Edenhofer, F.; Peitz, M.; Brustle, O. Stem cell engineering using transducible cre recombinase. Methods Mol. Med. 2007, 140, 17–32. [Google Scholar] [PubMed]
- Miyaoka, Y.; Chan, A.H.; Judge, L.M.; Yoo, J.; Huang, M.; Nguyen, T.D.; Lizarraga, P.P.; So, P.L.; Conklin, B.R. Isolation of single-base genome-edited human ips cells without antibiotic selection. Nat. Methods 2014, 11, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Thibaud, E.; Salomon-Bernard, Y.; Rappaport, R. Prepubertal genital hemorrhage. Study of 50 cases. Ann. Pediatr. (Paris) 1984, 31, 195–198. [Google Scholar] [PubMed]
- Bouhassira, E.E.; Westerman, K.; Leboulch, P. Transcriptional behavior of lcr enhancer elements integrated at the same chromosomal locus by recombinase-mediated cassette exchange. Blood 1997, 90, 3332–3344. [Google Scholar] [PubMed]
- Yang, L.; Guell, M.; Byrne, S.; Yang, J.L.; de Los Angeles, A.; Mali, P.; Aach, J.; Kim-Kiselak, C.; Briggs, A.W.; Rios, X.; et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013, 41, 9049–9061. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Simons, J.P.; Owen, J.S. Oligonucleotide-directed gene-editing technology: Mechanisms and future prospects. Expert Opin. Biol. Ther. 2012, 12, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Radecke, F.; Peter, I.; Radecke, S.; Gellhaus, K.; Schwarz, K.; Cathomen, T. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol. Ther. 2006, 14, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Pruett-Miller, S.M.; Huang, Y.; Gjoka, M.; Duda, K.; Taunton, J.; Collingwood, T.N.; Frodin, M.; Davis, G.D. High-frequency genome editing using ssdna oligonucleotides with zinc-finger nucleases. Nat. Methods 2011, 8, 753–755. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Laganiere, J.; Cheng, A.W.; Hockemeyer, D.; Gao, Q.; Alagappan, R.; Khurana, V.; Golbe, L.I.; Myers, R.H.; Lindquist, S.; et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset parkinson point mutations. Cell 2011, 146, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Zhang, S.C. Genetic modification of human embryonic stem cells. Biotechnol. Genet. Eng. Rev. 2007, 24, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, R. Lentivirus-mediated modification of pluripotent stem cells. Methods Mol. Biol. 2011, 767, 315–331. [Google Scholar] [PubMed]
- Siemen, H.; Nolden, L.; Terstegge, S.; Koch, P.; Brustle, O. Nucleofection of human embryonic stem cells. Methods Mol. Biol. 2008, 423, 131–138. [Google Scholar] [PubMed]
- Krentz, N.A.; Nian, C.; Lynn, F.C. TALEN/CRISPR-mediated eGFP knock-in add-on at the OCT4 locus does not impact differentiation of human embryonic stem cells towards endoderm. PLoS ONE 2014, 9, e114275. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human escs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Verma, N.; Gonzalez, F.; Shi, Z.D.; Huangfu, D. A CRISPR/Cas-mediated selection-free knockin strategy in human embryonic stem cells. Stem Cell Rep. 2015, 4, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Wang, H.; Kiani, S.; Lai, C.S.; Gao, Q.; Cassady, J.P.; Cost, G.J.; Zhang, L.; Santiago, Y.; Miller, J.C.; et al. Genetic engineering of human pluripotent cells using tale nucleases. Nat. Biotechnol. 2011, 29, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, R.; Chiba, K.; Schaeffer, L.; Regalado, S.G.; Lai, C.S.; Gao, Q.; Kiani, S.; Farin, H.F.; Clevers, H.; Cost, G.J.; et al. Human intestinal tissue with adult stem cell properties derived from pluripotent stem cells. Stem Cell Rep. 2014, 2, 838–852. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.W.; Haidet-Phillips, A.M.; Pham, J.T.; Lee, Y.; Huo, Y.; Tienari, P.J.; Maragakis, N.J.; Sattler, R.; Rothstein, J.D. Generation of gfap::Gfp astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia 2016, 64, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hunt, S.D.; Xue, H.; Liu, Y.; Darabi, R. Generation and validation of PAX7 reporter lines from human iPS cells using CRISPR/Cas9 technology. Stem Cell Res. 2016, 16, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Hunt, S.D.; Xue, H.; Liu, Y.; Darabi, R. Generation and characterization of a MYF5 reporter human iPS cell line using CRISPR/Cas9 mediated homologous recombination. Sci. Rep. 2016, 6, 18759. [Google Scholar] [CrossRef] [PubMed]
- DeKelver, R.C.; Choi, V.M.; Moehle, E.A.; Paschon, D.E.; Hockemeyer, D.; Meijsing, S.H.; Sancak, Y.; Cui, X.; Steine, E.J.; Miller, J.C.; et al. Functional genomics, proteomics, and regulatory DNA analysis in isogenic settings using zinc finger nuclease-driven transgenesis into a safe harbor locus in the human genome. Genome Res. 2010, 20, 1133–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tay, F.C.; Tan, W.K.; Goh, S.L.; Ramachandra, C.J.; Lau, C.H.; Zhu, H.; Chen, C.; Du, S.; Phang, R.Z.; Shahbazi, M.; et al. Targeted transgene insertion into the AAVS1 locus driven by baculoviral vector-mediated zinc finger nuclease expression in human-induced pluripotent stem cells. J. Gene Med. 2013, 15, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, W.Y.; Hu, S.; Lan, F.; Lee, A.S.; Huber, B.; Lisowski, L.; Liang, P.; Huang, M.; de Almeida, P.E.; et al. Genome editing of human embryonic stem cells and induced pluripotent stem cells with zinc finger nucleases for cellular imaging. Circ. Res. 2012, 111, 1494–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lau, C.H.; Goh, S.L.; Liang, Q.; Chen, C.; Du, S.; Phang, R.Z.; Tay, F.C.; Tan, W.K.; Li, Z.; et al. Baculoviral transduction facilitates TALEN-mediated targeted transgene integration and Cre/LoxP cassette exchange in human-induced pluripotent stem cells. Nucleic Acids Res. 2013, 41, e180. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, C.; Cerbini, T.; San, H.; Lin, Y.; Chen, G.; Rao, M.S.; Zou, J. Stable enhanced green fluorescent protein expression after differentiation and transplantation of reporter human induced pluripotent stem cells generated by AAVS1 transcription activator-like effector nucleases. Stem Cells Transl. Med. 2014, 3, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Mali, P.; Huang, X.; Dowey, S.N.; Cheng, L. Site-specific gene correction of a point mutation in human iPS cells derived from an adult patient with sickle cell disease. Blood 2011, 118, 4599–4608. [Google Scholar] [CrossRef] [PubMed]
- Sebastiano, V.; Maeder, M.L.; Angstman, J.F.; Haddad, B.; Khayter, C.; Yeo, D.T.; Goodwin, M.J.; Hawkins, J.S.; Ramirez, C.L.; Batista, L.F.; et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells 2011, 29, 1717–1726. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Dolatabadi, N.; Chan, S.F.; Zhang, X.; Akhtar, M.W.; Parker, J.; Soldner, F.; Sunico, C.R.; Nagar, S.; Talantova, M.; et al. Isogenic human iPSC Parkinson's model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 2013, 155, 1351–1364. [Google Scholar] [CrossRef] [PubMed]
- Fong, H.; Wang, C.; Knoferle, J.; Walker, D.; Balestra, M.E.; Tong, L.M.; Leung, L.; Ring, K.L.; Seeley, W.W.; Karydas, A.; et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Rep. 2013, 1, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef] [PubMed]
- Vossel, K.A.; Miller, B.L. New approaches to the treatment of frontotemporal lobar degeneration. Curr. Opin. Neurol. 2008, 21, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H.; et al. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015, 4, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maetzel, D.; Sarkar, S.; Wang, H.; Abi-Mosleh, L.; Xu, P.; Cheng, A.W.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from niemann-pick type c patient-specific iPS cells. Stem Cell Rep. 2014, 2, 866–880. [Google Scholar] [CrossRef] [PubMed]
- Frank, S.; Skryabin, B.V.; Greber, B. A modified TALEN-based system for robust generation of knock-out human pluripotent stem cell lines and disease models. BMC Genomics 2013, 14, 773. [Google Scholar] [CrossRef] [PubMed]
- Menon, T.; Firth, A.L.; Scripture-Adams, D.D.; Galic, Z.; Qualls, S.J.; Gilmore, W.B.; Ke, E.; Singer, O.; Anderson, L.S.; Bornzin, A.R.; et al. Lymphoid regeneration from gene-corrected SCID-X1 subject-derived iPSCs. Cell Stem Cell 2015, 16, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Zhao, H. Seamless correction of the sickle cell disease mutation of the hbb gene in human induced pluripotent stem cells using TALENs. Biotechnol. Bioeng. 2014, 111, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, H.; Kagoya, Y.; Kataoka, K.; Yoshimi, A.; Miyauchi, M.; Taoka, K.; Kumano, K.; Yamamoto, T.; Hotta, A.; Arai, S.; et al. Targeted gene correction of RUNX1 in induced pluripotent stem cells derived from familial platelet disorder with propensity to myeloid malignancy restores normal megakaryopoiesis. Exp. Hematol. 2015, 43, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Garate, Z.; Quintana-Bustamante, O.; Crane, A.M.; Olivier, E.; Poirot, L.; Galetto, R.; Kosinski, P.; Hill, C.; Kung, C.; Agirre, X.; et al. Generation of a high number of healthy erythroid cells from gene-edited pyruvate kinase deficiency patient-specific induced pluripotent stem cells. Stem Cell Rep. 2015, 5, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Kim, J.; Kweon, J.; Son, J.S.; Lee, J.S.; Yoo, J.E.; Cho, S.R.; Kim, J.H.; Kim, J.S.; Kim, D.W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. USA 2014, 111, 9253–9258. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Young, J.E.; Martinez, F.J.; Buen, F.; Gore, A.; Kinaga, J.; Li, Z.; Yuan, S.H.; Zhang, K.; Goldstein, L.S. The presenilin-1 DeltaE9 mutation results in reduced gamma-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell. Rep. 2013, 5, 974–985. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, G.; Reyna, S.M.; Dunlap, M.; van Der Kant, R.; Callender, J.A.; Young, J.E.; Roberts, E.A.; Goldstein, L.S. Defective transcytosis of app and lipoproteins in human iPSC-derived neurons with familial Alzheimer’s disease mutations. Cell Rep. 2016, 17, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Lane-Donovan, C.; Philips, G.T.; Herz, J. More than cholesterol transporters: Lipoprotein receptors in CNS function and neurodegeneration. Neuron 2014, 83, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Mauch, D.H.; Nagler, K.; Schumacher, S.; Goritz, C.; Muller, E.C.; Otto, A.; Pfrieger, F.W. CNS synaptogenesis promoted by glia-derived cholesterol. Science 2001, 294, 1354–1357. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Tyteca, D.; D'Auria, L.; Dewachter, I.; Gailly, P.; Hendrickx, A.; Tasiaux, B.; Haylani, L.E.; Muls, N.; N'Kuli, F.; et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol. Med. 2013, 5, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ye, L.; Chang, J.C.; Beyer, A.I.; Wang, J.; Muench, M.O.; Kan, Y.W. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014, 24, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Fan, Y.; He, W.; Zhu, D.; Niu, X.; Wang, D.; Ou, Z.; Luo, M.; Sun, X. Improved hematopoietic differentiation efficiency of gene-corrected beta-thalassemia induced pluripotent stem cells by CRISPR/Cas9 system. Stem Cells Dev. 2015, 24, 1053–1065. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional correction of large factor viii gene chromosomal inversions in hemophilia a patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Horii, T.; Tamura, D.; Morita, S.; Kimura, M.; Hatada, I. Generation of an icf syndrome model by efficient genome editing of human induced pluripotent stem cells using the CRISPR system. Int. J. Mol. Sci. 2013, 14, 19774–19781. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, Y.; Yan, W.; Smith, C.; Ye, Z.; Wang, J.; Gao, Y.; Mendelsohn, L.; Cheng, L. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 2015, 33, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Lai, Y.S.; Westin, E.; Khodadadi-Jamayran, A.; Pawlik, K.M.; Lamb, L.S., Jr.; Goldman, F.D.; Townes, T.M. Modeling human severe combined immunodeficiency and correction by CRISPR/Cas9-enhanced gene targeting. Cell. Rep. 2015, 12, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Menon, T.; Parker, G.S.; Qualls, S.J.; Lewis, B.M.; Ke, E.; Dargitz, C.T.; Wright, R.; Khanna, A.; Gage, F.H.; et al. Functional gene correction for cystic fibrosis in lung epithelial cells generated from patient iPSCs. Cell Rep. 2015, 12, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howden, S.E.; Maufort, J.P.; Duffin, B.M.; Elefanty, A.G.; Stanley, E.G.; Thomson, J.A. Simultaneous reprogramming and gene correction of patient fibroblasts. Stem Cell Rep. 2015, 5, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Yusa, K.; Rashid, S.T.; Strick-Marchand, H.; Varela, I.; Liu, P.Q.; Paschon, D.E.; Miranda, E.; Ordonez, A.; Hannan, N.R.; Rouhani, F.J.; et al. Targeted gene correction of alpha1-antitrypsin deficiency in induced pluripotent stem cells. Nature 2011, 478, 391–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kucherer, C.; Blau, O.; et al. Long-term control of hiv by CCR5 delta32/delta32 stem-cell transplantation. N Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y. CCR5-targeted hematopoietic stem cell gene approaches for hiv disease: Current progress and future prospects. Curr. Stem Cell. Res. Ther. 2012, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5delta32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Liu, Y.; Ma, T.; Liu, K.; Xu, S.; Zhang, Y.; Liu, H.; La Russa, M.; Xie, M.; Ding, S.; et al. Small molecules enhance CRISPR genome editing in pluripotent stem cells. Cell Stem Cell 2015, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Daer, R.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The impact of chromatin dynamics on Cas9-mediated genome editing in human cells. ACS Synth Biol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Westenskow, P.D.; Todorova, D.; Hu, Z.; Lin, T.; Rong, Z.; Kim, J.; He, J.; Wang, M.; et al. Humanized mice reveal differential immunogenicity of cells derived from autologous induced pluripotent stem cells. Cell Stem Cell 2015, 17, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Yu, M.; Shen, C.; Chen, X.; Feng, T.; Yao, Y.; Li, J.; Li, H.; Tu, W. Negligible immunogenicity of induced pluripotent stem cells derived from human skin fibroblasts. PLoS ONE 2014, 9, e114949. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, F.; Lei, J.; Fang, Y.; Tong, H.; Wu, W.; Liu, C. Pyrosequencing revealed highly microbial phylogenetic diversity in ferromanganese nodules from farmland. Environ. Sci. Process. Impacts 2015, 17, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Miyagawa, S.; Fukushima, S.; Maeda, A.; Kashiyama, N.; Kawamura, A.; Miki, K.; Okita, K.; Yoshida, Y.; Shiina, T.; et al. Cardiomyocytes derived from mhc-homozygous induced pluripotent stem cells exhibit reduced allogeneic immunogenicity in mhc-matched non-human primates. Stem Cell Rep. 2016, 6, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Rong, Z.; Wang, M.; Hu, Z.; Stradner, M.; Zhu, S.; Kong, H.; Yi, H.; Goldrath, A.; Yang, Y.G.; Xu, Y.; et al. An effective approach to prevent immune rejection of human esc-derived allografts. Cell Stem Cell 2014, 14, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Shen, X.Z.; Jiang, F.; Wu, Y.; Han, C. DNA-guided genome editing using the natronobacterium gregoryi argonaute. Nat. Biotechnol. 2016, 34, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Dong, Z.; Shi, Y.; Wang, X.; Qin, Y.; Wang, Y.; Liu, D. NgAgo-based fabp11a gene knockdown causes eye developmental defects in zebrafish. Cell Res. 2016, 26, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Updated: NgAgo gene-editing controversy escalates in peer-reviewed papers. Nature 2016, 540, 20–21. [Google Scholar] [CrossRef] [PubMed]
- Cyranoski, D. Replications, ridicule and a recluse: The controversy over ngago gene-editing intensifies. Nature 2016, 536, 136–137. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Cheng, L.; Gu, F.; Huang, J.; Huang, Z.; Lin, S.; Li, J.; Li, W.; Qin, W.; Sun, Y.; et al. Questions about ngago. Protein Cell 2016, 7, 913–915. [Google Scholar] [CrossRef] [PubMed]
- Sandoe, J.; Eggan, K. Opportunities and challenges of pluripotent stem cell neurodegenerative disease models. Nat. Neurosci. 2013, 16, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.E.; Dai, D.F.; Laflamme, M.A. Human pluripotent stem cells: Prospects and challenges as a source of cardiomyocytes for in vitro modeling and cell-based cardiac repair. Adv. Drug Deliv. Rev. 2016, 96, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Ding, S.; Rando, T.A.; Trounson, A. Translational strategies and challenges in regenerative medicine. Nat. Med. 2014, 20, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Trounson, A.; DeWitt, N.D. Pluripotent stem cells progressing to the clinic. Nat. Rev. Mol. Cell Biol. 2016, 17, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Picanco-Castro, V.; Moreira, L.F.; Kashima, S.; Covas, D.T. Can pluripotent stem cells be used in cell-based therapy? Cell Reprogram 2014, 16, 98–107. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cas9 Variants | Advantages | Disadvantages |
|---|---|---|
| Wild type Streptococcus pyogenes Cas9 (spCas9) | Programmed RNA guided editing; High specificity; Easily engineered | dsDNA breaks repaired by NHEJ forming indels |
| Cas9 nickase (Cas9n) | No dsDNA break induced; Promotes homology directed repair (HDR) | Some nicks go through a dsDNA break intermediate that can be repaired by NHEJ |
| Dual sg-RNA-Cas9 nickases (Cas9dn) | Increased specificity, dual sgRNA, promotes higher HDR over single nickase. | Must design dual sg-RNA-Cas9n complexes targeting opposite DNA strands |
| Cytidine deaminase fused Cas9 (D10A) | No dsDNA break induced; Increased efficiency over spCas9; Direct base conversion of C→T | Five base pair editing window; specific C→T conversion |
| spCas9-Gem | Regulates Cas9 presence at each stage of the cell cycle; Efficiently generates knock-in reporter lines and gene correction | Decreases frequency of NHEJ indels at target locus |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brookhouser, N.; Raman, S.; Potts, C.; Brafman, D.A. May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells. Cells 2017, 6, 5. https://doi.org/10.3390/cells6010005
Brookhouser N, Raman S, Potts C, Brafman DA. May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells. Cells. 2017; 6(1):5. https://doi.org/10.3390/cells6010005
Chicago/Turabian StyleBrookhouser, Nicholas, Sreedevi Raman, Christopher Potts, and David. A. Brafman. 2017. "May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells" Cells 6, no. 1: 5. https://doi.org/10.3390/cells6010005
APA StyleBrookhouser, N., Raman, S., Potts, C., & Brafman, D. A. (2017). May I Cut in? Gene Editing Approaches in Human Induced Pluripotent Stem Cells. Cells, 6(1), 5. https://doi.org/10.3390/cells6010005