Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Tissue Samples and Cell Culture
2.2. Histone Protein Extraction
2.3. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and In-Gel Digestion
2.4. Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
2.5. Workflow of Wide Tolerance Acetylation Analysis
2.6. Production of Anti-H3T22ac Antibody
2.7. Western Blot Analysis
3. Results and Discussion
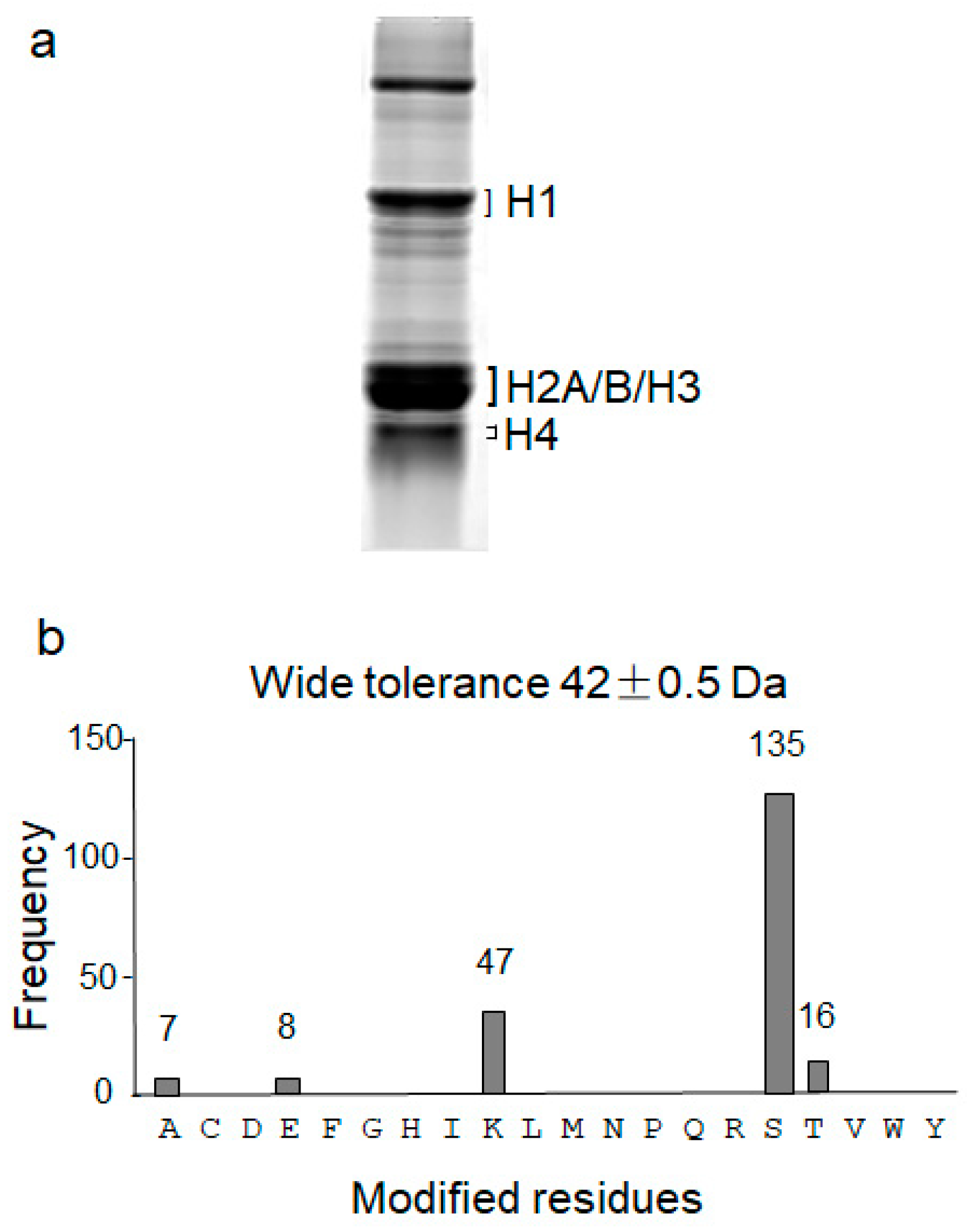
3.1. High Peptide Coverage of Human Histones
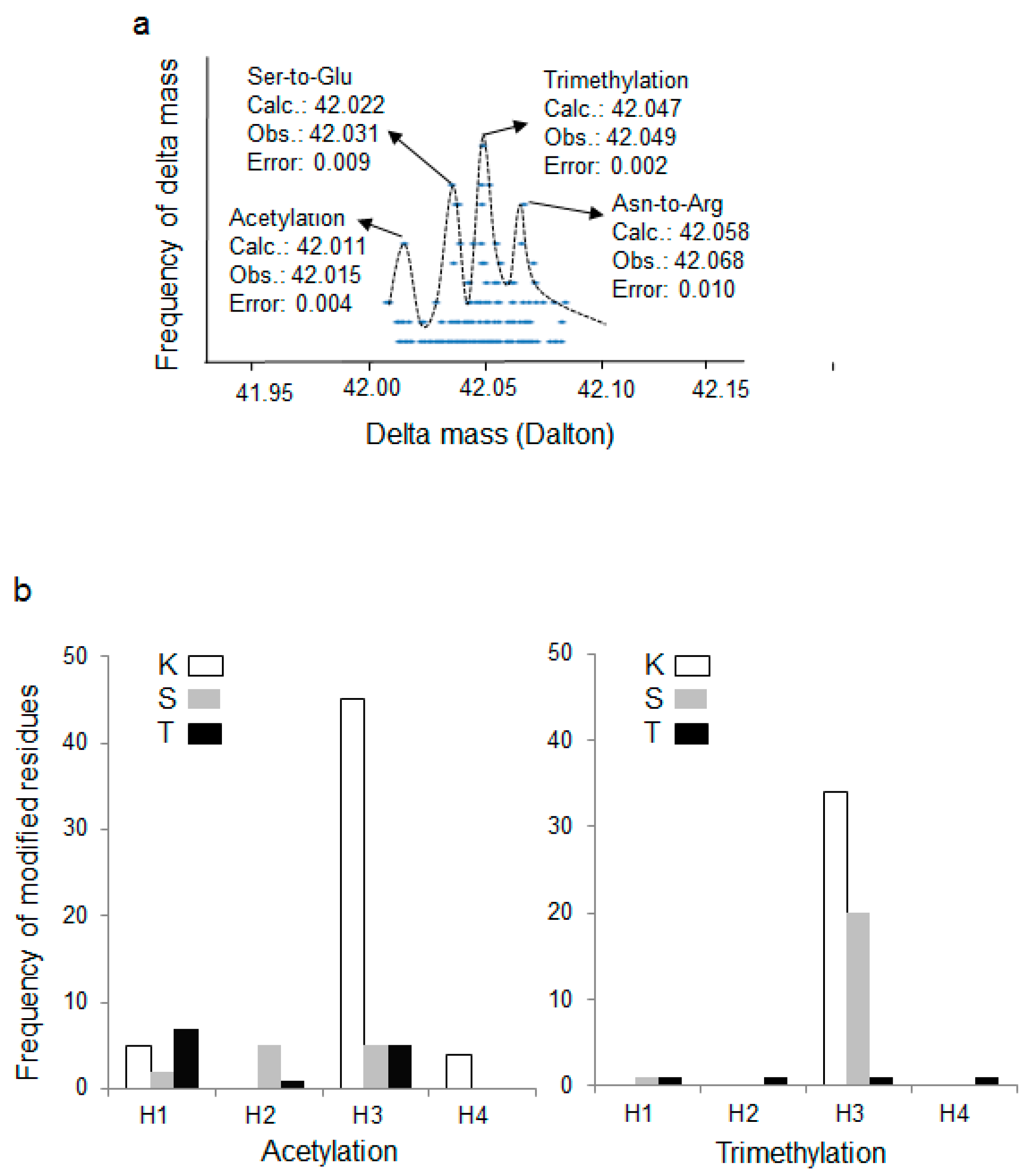
3.2. Wide Tolerance Acetylation Analysis of Human Histones at Undefined Amino Acid Residues
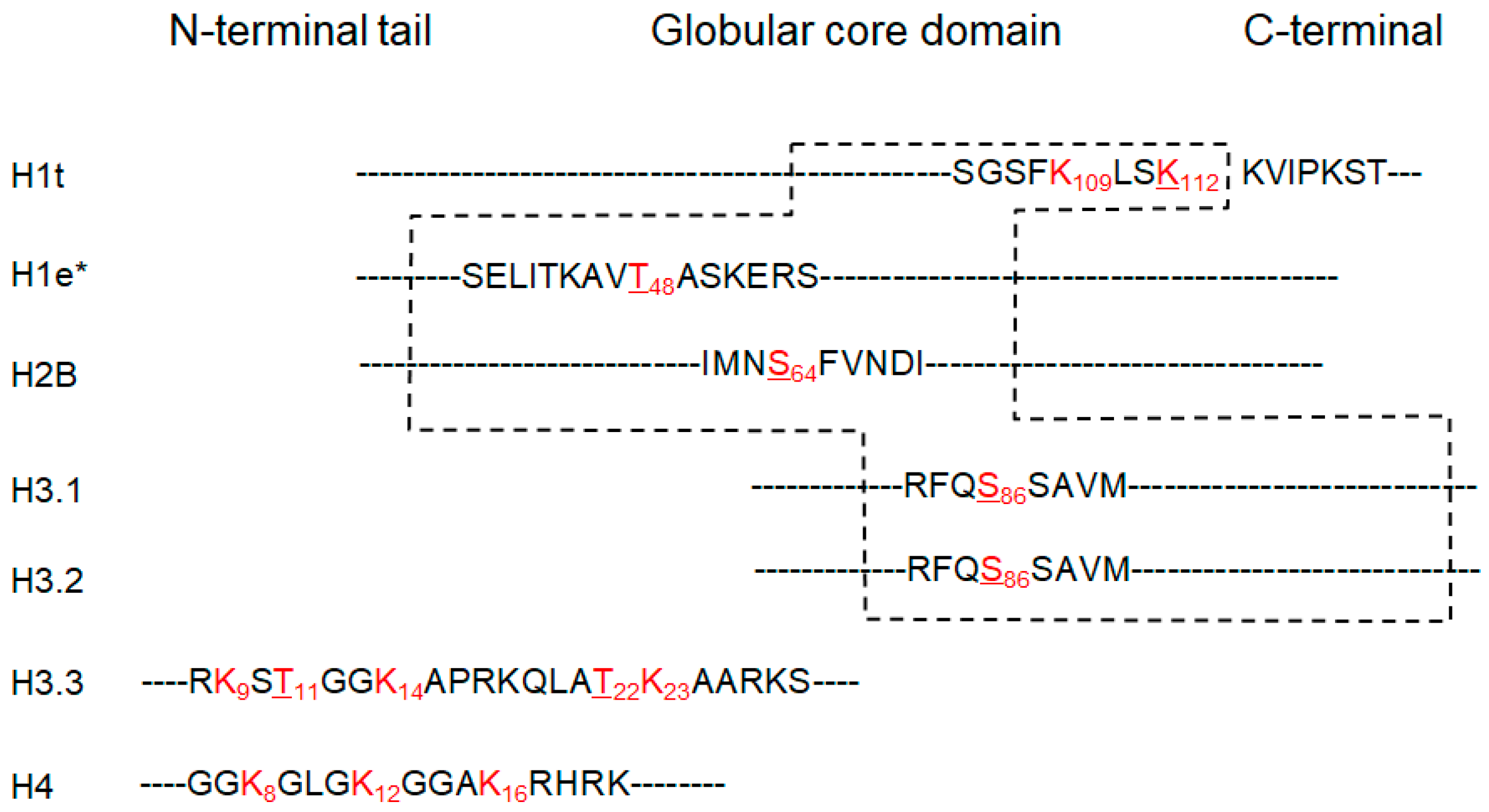
3.3. Putative New Acetylation Modifications on Human Histones by Restricted Delta Mass Search
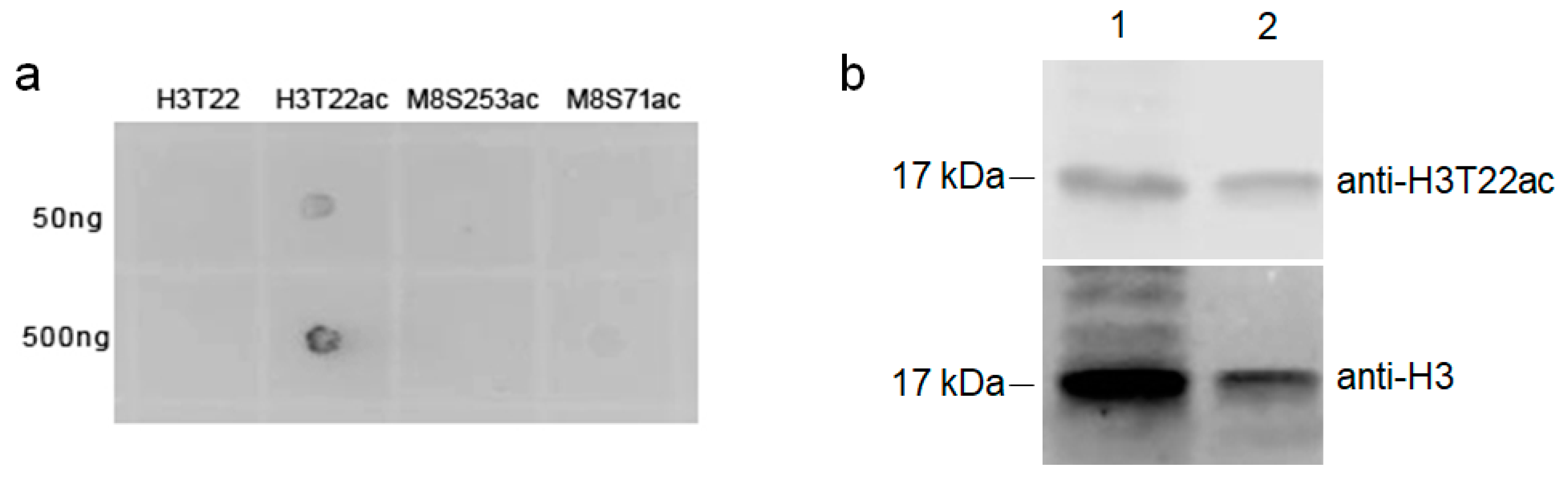
3.4. Validation of T-Acetylation in the N-Terminal Tail of Human Histone H3 (H3T22ac) by Immune Assays
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Shogren-Knaak, M.; Ishii, H.; Sun, J.M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- Rundlett, S.E.; Carmen, A.A.; Suka, N.; Turner, B.M.; Grunstein, M. Transcriptional repression by UME6 involves deacetylation of lysine 5 of histone H4 by RPD3. Nature 1998, 392, 831–835. [Google Scholar] [PubMed]
- Patel, D.J.; Wang, Z. Readout of epigenetic modifications. Annu. Rev. Biochem. 2013, 82, 81–118. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat. Struct. Mol. Biol. 2005, 12, 110–112. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; North, J.A.; Shimko, J.C.; Forties, R.A.; Ferdinand, M.B.; Manohar, M.; Zhang, M.; Fishel, R.; Ottesen, J.J.; Poirier, M.G. Histone fold modifications control nucleosome unwrapping and disassembly. Proc. Natl. Acad. Sci. USA 2011, 108, 12711–12716. [Google Scholar] [CrossRef] [PubMed]
- Hyland, E.M.; Cosgrove, M.S.; Molina, H.; Wang, D.; Pandey, A.; Cottee, R.J.; Boeke, J.D. Insights into the role of histone H3 and histone H4 core modifiable residues in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005, 25, 10060–10070. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Zhang, K.; Grunstein, M. Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 2005, 121, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Carson, J.J.; Feser, J.; Tamburini, B.; Zabaronick, S.; Linger, J.; Tyler, J.K. Acetylated lysine 56 on histone H3 drives chromatin assembly after repair and signals for the completion of repair. Cell 2008, 134, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Truong, D.; Tyler, J.K. Acetylation in the globular core of histone H3 on lysine-56 promotes chromatin disassembly during transcriptional activation. Proc. Natl. Acad. Sci. USA 2008, 105, 9000–9005. [Google Scholar] [CrossRef] [PubMed]
- Manohar, M.; Mooney, A.M.; North, J.A.; Nakkula, R.J.; Picking, J.W.; Edon, A.; Fishel, R.; Poirier, M.G.; Ottesen, J.J. Acetylation of histone H3 at the nucleosome dyad alters DNA-histone binding. J. Biol. Chem. 2009, 284, 23312–23321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Eugeni, E.E.; Parthun, M.R.; Freitas, M.A. Identification of novel histone post-translational modifications by peptide mass fingerprinting. Chromosoma 2003, 112, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Schiza, V.; Molina-Serrano, D.; Kyriakou, D.; Hadjiantoniou, A.; Kirmizis, A. N-alpha-terminal acetylation of histone H4 regulates arginine methylation and ribosomal DNA silencing. PLoS Genet. 2013, 9, e1003805. [Google Scholar] [CrossRef] [PubMed]
- Starheim, K.K.; Gevaert, K.; Arnesen, T. Protein N-terminal acetyltransferases: When the start matters. Trends Biochem. Sci. 2012, 37, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Hell, R. Molecular physiology of plant sulfur metabolism. Planta 1997, 202, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Peak-Chew, S.Y.; McMahon, H.T. Acetylation of MEK2 and I kappa B kinase (IKK) activation loop residues by YopJ inhibits signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18574–18579. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Keitany, G.; Li, Y.; Wang, Y.; Ball, H.L.; Goldsmith, E.J.; Orth, K. Yersinia YopJ acetylates and inhibits kinase activation by blocking phosphorylation. Science 2006, 312, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Shechter, D.; Dormann, H.L.; Allis, C.D.; Hake, S.B. Extraction, purification and analysis of histones. Nat. Protoc. 2007, 2, 1445–1457. [Google Scholar] [CrossRef] [PubMed]
- Na, S.; Bandeira, N.; Paek, E. Fast multi-blind modification search through tandem mass spectrometry. Mol. Cell. Proteom. 2012, 11, M111.010199. [Google Scholar] [CrossRef] [PubMed]
- Chick, J.M.; Kolippakkam, D.; Nusinow, D.P.; Zhai, B.; Rad, R.; Huttlin, E.L.; Gygi, S.P. A mass-tolerant database search identifies a large proportion of unassigned spectra in shotgun proteomics as modified peptides. Nat. Biotechnol. 2015, 33, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [PubMed]
- Bonissone, S.; Gupta, N.; Romine, M.; Bradshaw, R.A.; Pevzner, P.A. N-terminal protein processing: A comparative proteogenomic analysis. Mol. Cell. Proteom. 2013, 12, 14–28. [Google Scholar] [CrossRef] [PubMed]
- Giglione, C.; Boularot, A.; Meinnel, T. Protein N-terminal methionine excision. Cell. Mol. Life Sci. 2004, 61, 1455–1474. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M.; Grimes, G.R.; Kumar, Y.; Olley, G.; Taylor, G.C.; Schneider, R.; Bickmore, W.A. Histone H3 globular domain acetylation identifies a new class of enhancers. Nat. Genet. 2016, 48, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Szenker, E.; Ray-Gallet, D.; Almouzni, G. The double face of the histone variant H3.3. Cell Res. 2011, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Li, S.; Chavez, V.; Lanting, L.; Natarajan, R. Coactivator-associated arginine methyltransferase-1 enhances nuclear factor-kappaB-mediated gene transcription through methylation of histone H3 at arginine 17. Mol. Endocrinol. 2006, 20, 1562–1573. [Google Scholar] [CrossRef] [PubMed]
- Daujat, S.; Bauer, U.M.; Shah, V.; Turner, B.; Berger, S.; Kouzarides, T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 2002, 12, 2090–2097. [Google Scholar] [CrossRef]
- Britton, L.M.; Newhart, A.; Bhanu, N.V.; Sridharan, R.; Gonzales-Cope, M.; Plath, K.; Janicki, S.M.; Garcia, B.A. Initial characterization of histone H3 serine 10 O-acetylation. Epigenetics 2013, 8, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Tweedie-Cullen, R.Y.; Brunner, A.M.; Grossmann, J.; Mohanna, S.; Sichau, D.; Nanni, P.; Panse, C.; Mansuy, I.M. Identification of combinatorial patterns of post-translational modifications on individual histones in the mouse brain. PLoS ONE 2012, 7, e36980. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Choi, H.-P.; Wang, X.; Wu, F.; Chen, X.; Lü, X.; Jing, R.; Ryu, H.; Wang, X.; Azadzoi, K.M.; et al. Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation. Cells 2017, 6, 34. https://doi.org/10.3390/cells6040034
Li C, Choi H-P, Wang X, Wu F, Chen X, Lü X, Jing R, Ryu H, Wang X, Azadzoi KM, et al. Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation. Cells. 2017; 6(4):34. https://doi.org/10.3390/cells6040034
Chicago/Turabian StyleLi, Cuiling, Han-Pil Choi, Xiaoyue Wang, Fei Wu, Xinjun Chen, Xin Lü, Ruirui Jing, Hoon Ryu, Xingyuan Wang, Kazem M. Azadzoi, and et al. 2017. "Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation" Cells 6, no. 4: 34. https://doi.org/10.3390/cells6040034
APA StyleLi, C., Choi, H. -P., Wang, X., Wu, F., Chen, X., Lü, X., Jing, R., Ryu, H., Wang, X., Azadzoi, K. M., & Yang, J. -H. (2017). Post-Translational Modification of Human Histone by Wide Tolerance of Acetylation. Cells, 6(4), 34. https://doi.org/10.3390/cells6040034