Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle



Abstract
:1. Introduction
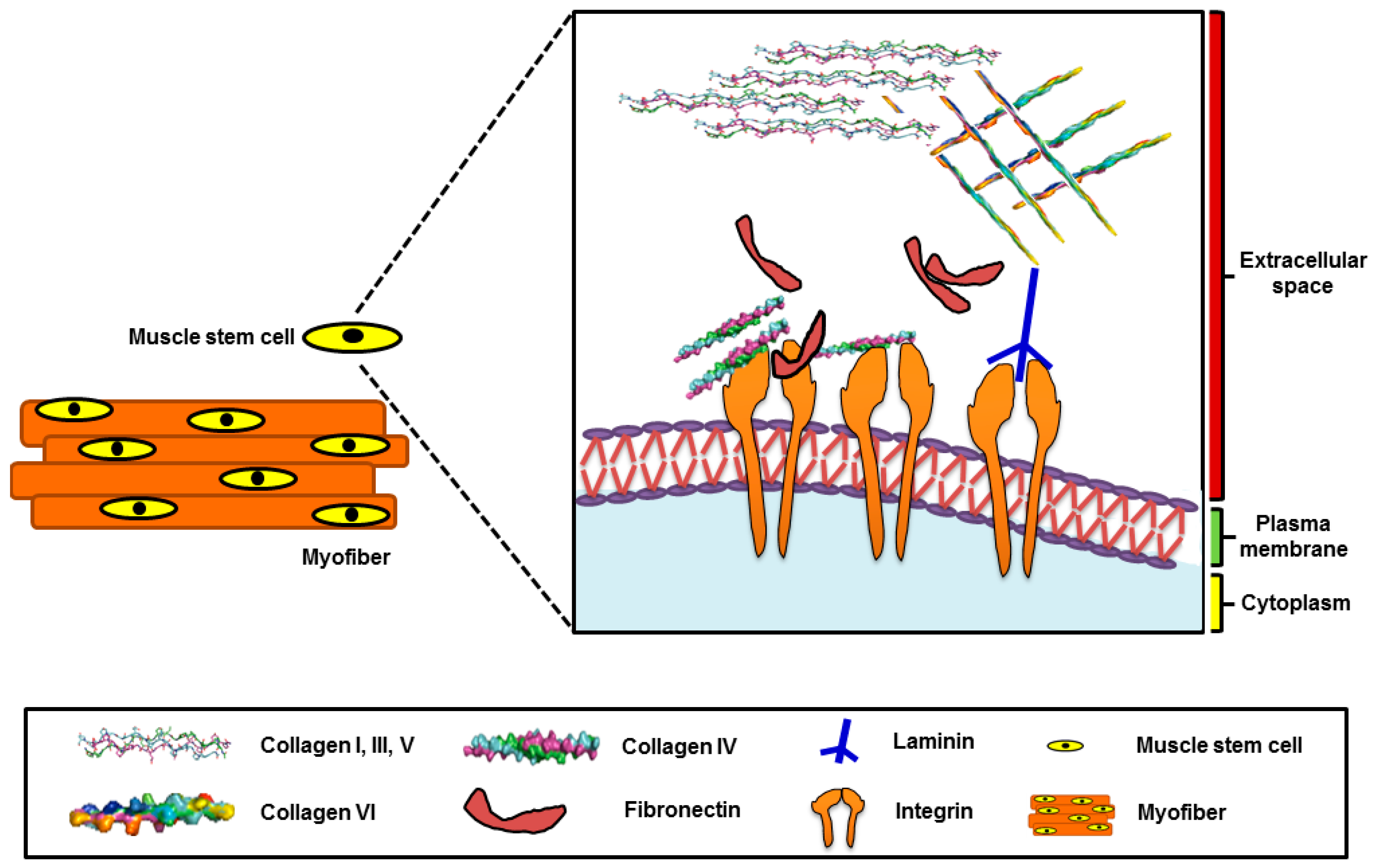
2. Extracellular Matrix
Muscle Stem Cells and Extracellular Matrix(ECM)
3. Insulin Resistance in Skeletal Muscle
4. The Extracellular Matrix (ECM) and Insulin Resistance
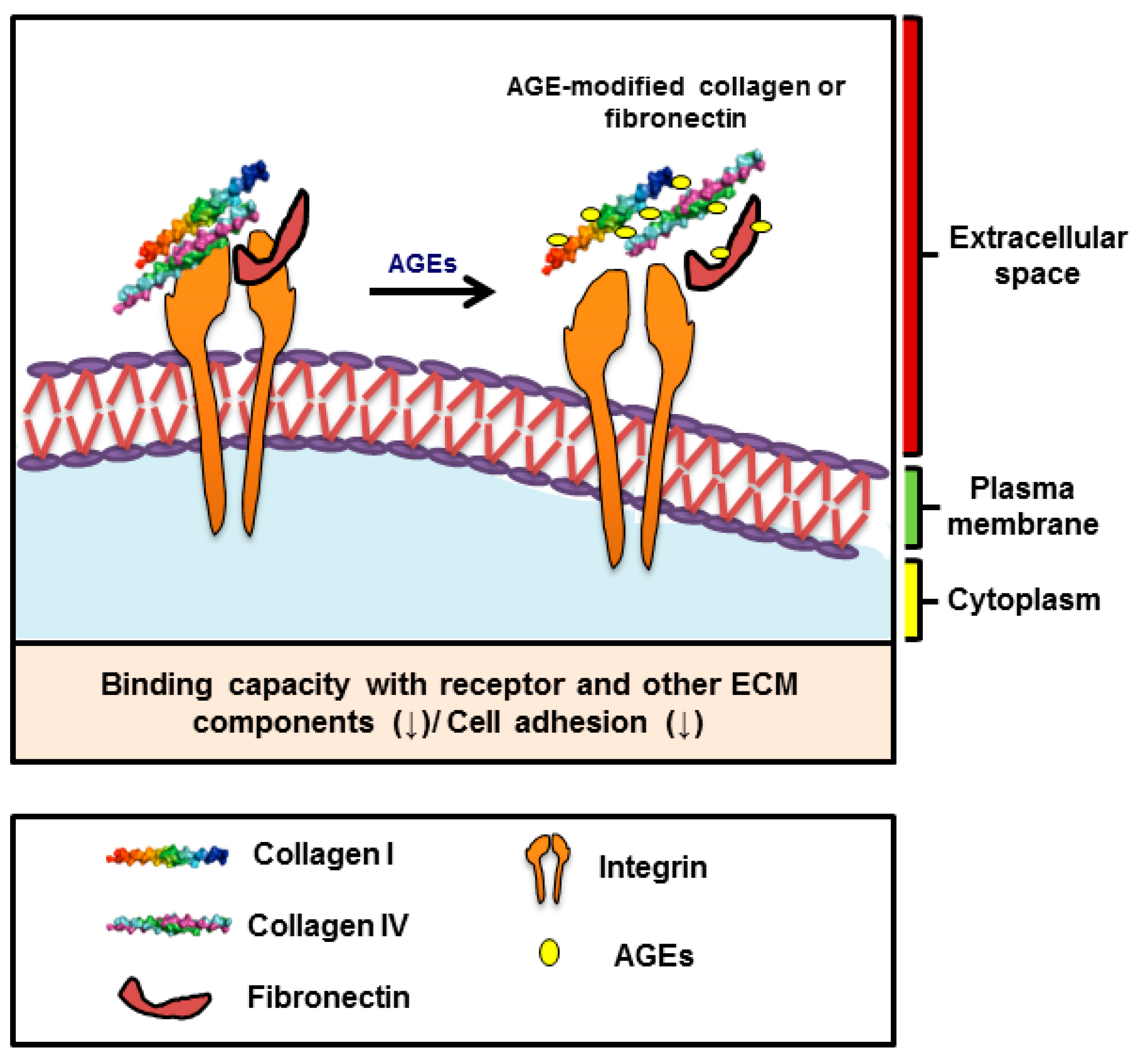
5. Glycation of Skeletal Muscle ECM
6. Insulin Resistance and Skeletal Muscle ECM Remodeling: Clinical Studies
7. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MSCs | muscle stem cells |
| GLUT4 | glucose transporter type 4 |
| IR | insulin resistance |
| T2DM | type 2 diabetes mellitus |
| ECM | extracellular matrix |
| FN | fibronectin |
| MFRs | muscle regulatory factors |
| HGF | hepatocyte growth factor |
| NOS | neuronal nitric oxide synthase |
| BM | basement membrane |
| BL | basal lamina |
| AMPK | AMP-activated protein kinase |
| GWAS | genome-wide association studies |
| ANK1 | ankyrin-1 |
| IRS | insulin receptor substrate-1 |
| PFKM | phosphofructokinase |
| ER | endoplasmic reticulum |
| IRS | insulin receptor substrate |
| IRE-1 | inositol-requiring enzyme 1 |
| TRB3 | ER stress upraises tribbles 3 |
| HFD | high-fat diet |
| PTP1B | protein tyrosine phosphatase 1B |
| SKIP | inositol polyphosphate phosphatase |
| TGs | triglycerides |
| MMP | matrix metalloproteases |
| FMOD | fibromodulin |
| MGP | matrix gla protein |
| MSTN | myostatin |
| ACVRIIB | activin receptor type II B |
| CTGF | connective tissue growth factor |
| MTJ | myotendinous |
| HA | hyaluronan |
| TGF | transforming growth factor |
| AGEs | advanced glycation end products |
References
- Kim, K.M.; Jang, H.C.; Lim, S. Differences among skeletal muscle mass indices derived from height-, weight-, and body mass index-adjusted models in assessing sarcopenia. Korean J. Intern. Med. 2016, 31, 643. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef] [PubMed]
- Shadrin, I.; Khodabukus, A.; Bursac, N. Striated muscle function, regeneration, and repair. Cell. Mol. Life Sci. 2016, 73, 4175–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiol. Rev. 2013, 93, 993–1017. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J. The effect of insulin on the disposal of intravenous glucose: Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; de Strijcker, D.; Calders, P. Impact of endurance exercise training in the fasted state on muscle biochemistry and metabolism in healthy subjects: Can these effects be of particular clinical benefit to type 2 diabetes mellitus and insulin-resistant patients? Sports Med. 2017, 47, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E. Is insulin resistance the principal cause of type 2 diabetes? Diabetes Obes. Metab. 1999, 1, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ahmad, K.; Malik, A.; Rabbani, G.; Kim, T.; Lee, I.-K.; Lee, Y.H.; Park, S.-Y.; et al. Fibromodulin and regulation of the intricate balance between myoblast differentiation to myocytes or adipocyte-like cells. FASEB J. 2017, 32, 768–781. [Google Scholar] [CrossRef] [PubMed]
- Asakura, A.; Rudnicki, M.A.; Komaki, M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic, and adipogenic differentiation. Differentiation 2001, 68, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, B. Biology of the extracellular matrix: An overview. J. Glaucoma 2014, 23, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Ayala, J.E.; Lee-Young, R.S.; Zhang, Z.; James, F.D.; Neufer, P.D.; Pozzi, A.; Zutter, M.M.; Wasserman, D.H. Diet-induced muscle insulin resistance is associated with extracellular matrix remodeling and interaction with integrin alpha2beta1 in mice. Diabetes 2011, 60, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.S.; Kang, L.; Wasserman, D.H. The extracellular matrix and insulin resistance. Trends Endocrinol. Metab. 2015, 26, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Bastie, C.C.; Xu, J.; Fassler, R.; Campbell, K.P.; Kurland, I.J.; Pessin, J.E. Insulin resistance in striated muscle-specific integrin receptor β1-deficient mice. J. Biol. Chem. 2009, 284, 4679–4688. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K. Hyperinsulinemic-euglycemic clamp to assess insulin sensitivity in vivo. Methods Mol. Biol. 2009, 560, 221–238. [Google Scholar] [PubMed]
- Martinez-Huenchullan, S.; McLennan, S.; Verhoeven, A.; Twigg, S.; Tam, C. The emerging role of skeletal muscle extracellular matrix remodelling in obesity and exercise. Obes. Rev. 2017, 18, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, P.; Riso, E.M.; Seene, T. Extracellular matrix and myofibrils during unloading and reloading of skeletal muscle. Intern. J. Sports Med. 2011, 32, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.C.; Mueller, U.; Conti, F.J. Integrins in the Development and Pathology of Skeletal Muscle. In Neuromuscular Disorders; InTech: London, UK, 2012; Available online: https://www.intechopen.com/books/neuromuscular-disorders/integrins-in-the-development-and-pathology-of-skeletal-muscle (accessed on 13 May 2018).[Green Version]
- Zaidel-Bar, R.; Itzkovitz, S.; Ma’ayan, A.; Iyengar, R.; Geiger, B. Functional atlas of the integrin adhesome. Nat. Cell Biol. 2007, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Cruz, M.; Sanchez, R.; Escobar, R.E.; Cruz-Guzmán, O.d.R.; López-Alarcón, M.; Bernabe;a, M.; Coral-Vázquez, R.; Matute, G.; Velázquez Wong, A.C. Evidence of insulin resistance and other metabolic alterations in boys with duchenne or becker muscular dystrophy. Inter. J. Endocrinol. 2015, 2015, 867273. [Google Scholar]
- Ricard-Blum, S.; Ruggiero, F.; van der Rest, M. The Collagen Superfamily. In Collagen; Springer: Berlin/Heidelberg, Germany, 2005; pp. 35–84. [Google Scholar]
- Bella, J.; Hulmes, D.J. Fibrillar collagens. In Fibrous Proteins: Structures and Mechanisms; Springer: Berlin/Heidelberg, Germany, 2017; pp. 457–490. [Google Scholar]
- Gelse, K.; Pöschl, E.; Aigner, T. Collagens—Structure, function, and biosynthesis. Adv. Drug Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huijing, P.A. Muscle as a collagen fiber reinforced composite: A review of force transmission in muscle and whole limb. J. Biomech. 1999, 32, 329–345. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Ruggiero, F. The collagen superfamily: From the extracellular matrix to the cell membrane. Pathol. Biol. 2005, 53, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Orgel, J.; San Antonio, J.; Antipova, O. Molecular and structural mapping of collagen fibril interactions. Connect. Tissue Res. 2011, 52, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Tam, E.M.; Wu, Y.I.; Butler, G.S.; Stack, M.S.; Overall, C.M. Collagen binding properties of the membrane type-1 matrix metalloproteinase (MT1-MMP) hemopexin c domain the ectodomain of the 44-kDa autocatalytic product of MT1-MMP inhibits cell invasion by disrupting native type I collagen cleavage. J. Biol. Chem. 2002, 277, 39005–39014. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.M.; Orgel, J.P.; Fertala, A.; McAuliffe, J.D.; Turner, K.R.; Di Lullo, G.A.; Chen, S.; Antipova, O.; Perumal, S.; Ala-Kokko, L. Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J. Biol. Chem. 2008, 283, 21187–21197. [Google Scholar] [CrossRef] [PubMed]
- Lukjanenko, L.; Jung, M.J.; Hegde, N.; Perruisseau-Carrier, C.; Migliavacca, E.; Rozo, M.; Karaz, S.; Jacot, G.; Schmidt, M.; Li, L.; et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 2016, 22, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, A.T.; Lee, E.J.; Choi, I. Fibromodulin: A regulatory molecule maintaining cellular architecture for normal cellular function. Int. J. Biochem. Cell. Biol. 2016, 80, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ashraf, J.M.; Nahm, S.S.; Kim, Y.W.; Park, S.Y.; Choi, I. Fibromodulin: A master regulator of myostatin controlling progression of satellite cells through a myogenic program. FASEB J. 2016, 30, 2708–2719. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Jan, A.T.; Baig, M.H.; Lee, E.J.; Choi, I. Matrix gla protein: An extracellular matrix protein regulates myostatin expression in the muscle developmental program. Life Sci. 2017, 172, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Covington, J.D.; Bajpeyi, S.; Tchoukalova, Y.; Burk, D.; Johannsen, D.L.; Zingaretti, C.M.; Cinti, S.; Ravussin, E. Weight gain reveals dramatic increases in skeletal muscle extracellular matrix remodeling. J. Clin. Endocrinol. Met. 2014, 99, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.C.; Marcus-Soekarman, D.; Stolte-Dijkstra, I.; Verrips, A.; Taylor, J.A.; Briggs, M.D. Type ix collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am. J. Med. Genet. A 2010, 152A, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Chiquet, M.; Birk, D.E.; Bonnemann, C.G.; Koch, M. Collagen xii: Protecting bone and muscle integrity by organizing collagen fibrils. Int. J. Biochem. Cell Biol. 2014, 53, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Listrat, A.; Pissavy, A.-L.; Micol, D.; Jurie, C.; Lethias, C.; Pethick, D.; Hocquette, J.-F. Collagens xii and xiv: Two collagen types both associated with bovine muscle and intramuscular lipid metabolism. Livest. Sci. 2016, 187, 80–86. [Google Scholar] [CrossRef]
- Eklund, L.; Piuhola, J.; Komulainen, J.; Sormunen, R.; Ongvarrasopone, C.; Fassler, R.; Muona, A.; Ilves, M.; Ruskoaho, H.; Takala, T.E.; et al. Lack of type XV collagen causes a skeletal myopathy and cardiovascular defects in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1194–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Law, B.; Fowlkes, V.; Goldsmith, J.G.; Carver, W.; Goldsmith, E.C. Diabetes-induced alterations in the extracellular matrix and their impact on myocardial function. Microsc. Microanal. 2012, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Gullberg, D.; Velling, T.; Lohikangas, L.; Tiger, C.F. Integrins during muscle development and in muscular dystrophies. Front. Biosci. 1998, 3, D1039–D1050. [Google Scholar] [CrossRef] [PubMed]
- Danoviz, M.E.; Yablonka-Reuveni, Z. Skeletal muscle satellite cells: Background and methods for isolation and analysis in a primary culture system. In Myogenesis; Springer: Berlin/Heidelberg, Germany, 2012; pp. 21–52. [Google Scholar]
- Fu, X.; Wang, H.; Hu, P. Stem cell activation in skeletal muscle regeneration. Cell. Mol. Life Sci. 2015, 72, 1663–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.; Engler, A.J.; Meyer, G.A. Extracellular matrix regulation in the muscle satellite cell niche. Connect. Tissue Res. 2015, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Niu, A.; Chen, S.E.; Li, Y.P. Beta3-integrin mediates satellite cell differentiation in regenerating mouse muscle. FASEB J. 2011, 25, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Sinha, I.; Sakthivel, D.; Varon, D.E. Systemic regulators of skeletal muscle regeneration in obesity. Front. Endocrinol. 2017, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.P.; Fisher, J.S. Skeletal muscle insulin resistance: Roles of fatty acid metabolism and exercise. Phys. Ther. 2008, 88, 1279–1296. [Google Scholar] [CrossRef] [PubMed]
- Keske, M.A.; Premilovac, D.; Bradley, E.A.; Dwyer, R.M.; Richards, S.M.; Rattigan, S. Muscle microvascular blood flow responses in insulin resistance and ageing. J. Physiol. 2016, 594, 2223–2231. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Lai, S.; Yang, Y.; Shi, H.; Cai, Z.; Sorrentino, V.; Du, H.; Chen, H. A novel type 2 diabetes risk allele increases the promoter activity of the muscle-specific small ankyrin 1 gene. Sci. Rep. 2016, 6, 25105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almind, K.; Inoue, G.; Pedersen, O.; Kahn, C.R. A common amino acid polymorphism in insulin receptor substrate-1 causes impaired insulin signaling. Evidence from transfection studies. J. Clin. Invest. 1996, 97, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Salvado, L.; Palomer, X.; Barroso, E.; Vazquez-Carrera, M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol. Metab. 2015, 26, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1b and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Ijuin, T.; Hosooka, T.; Takenawa, T. Phosphatidylinositol 3,4,5-trisphosphate phosphatase skip links endoplasmic reticulum stress in skeletal muscle to insulin resistance. Mol. Cell. Biol. 2016, 36, 108–118. [Google Scholar] [PubMed]
- Bonen, A.; Holloway, G.P.; Tandon, N.N.; Han, X.X.; McFarlan, J.; Glatz, J.F.; Luiken, J.J. Cardiac and skeletal muscle fatty acid transport and transporters and triacylglycerol and fatty acid oxidation in lean and zucker diabetic fatty rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D. Fatty acid oxidation and its relation with insulin resistance and associated disorders. Ann. Nutr. Metab. 2016, 68 (Suppl. 3), 15–20. [Google Scholar] [CrossRef]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.; Jeneson, J.A.; Backes, W.H.; van Echteld, C.J.; van Engelshoven, J.M.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and bmi-matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Holloway, G.P.; Thrush, A.B.; Heigenhauser, G.J.; Tandon, N.N.; Dyck, D.J.; Bonen, A.; Spriet, L.L. Skeletal muscle mitochondrial FAT/CD36 content and palmitate oxidation are not decreased in obese women. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1782–E1789. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Min, K.; Zhang, L.; Canfran-Duque, A.; Jurczak, M.J.; Camporez, J.P.G.; Nie, Y.; Gavin, T.P.; Shulman, G.I.; Hernandez-Fernando, C. Skeletal muscle-specific deletion of MKP-1 reveals a p38 MAPK/JNK/Akt signaling node that regulates obesity-induced insulin resistance. Diabetes 2018, 67, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Park, S.M. Association of muscle mass and fat mass with insulin resistance and the prevalence of metabolic syndrome in korean adults: A cross-sectional study. Sci. Rep. 2018, 8, 2703. [Google Scholar] [CrossRef] [PubMed]
- Berria, R.; Wang, L.; Richardson, D.K.; Finlayson, J.; Belfort, R.; Pratipanawatr, T.; De Filippis, E.A.; Kashyap, S.; Mandarino, L.J. Increased collagen content in insulin-resistant skeletal muscle. Am. J. Physiol.-Endocrinol. Met. 2006, 290, E560–E565. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.K.; Kashyap, S.; Bajaj, M.; Cusi, K.; Mandarino, S.J.; Finlayson, J.; DeFronzo, R.A.; Jenkinson, C.P.; Mandarino, L.J. Lipid infusion decreases the expression of nuclear encoded mitochondrial genes and increases the expression of extracellular matrix genes in human skeletal muscle. J. Biol. Chem. 2005, 280, 10290–10297. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Chaudhuri, R.; Hutchison, A.T.; Samocha-Bonet, D.; Heilbronn, L.K. Skeletal muscle extracellular matrix remodeling after short-term overfeeding in healthy humans. Metabolism 2017, 67, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Mayes, W.H.; James, F.D.; Bracy, D.P.; Wasserman, D.H. Matrix metalloproteinase 9 opposes diet-induced muscle insulin resistance in mice. Diabetologia 2014, 57, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox. Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [PubMed]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGF β1 on ROS generators, mediators and functional consequences. Redox. Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, O.; Severcan, M.; Severcan, F. Diabetes induces compositional, structural and functional alterations on rat skeletal soleus muscle revealed by ftir spectroscopy: A comparative study with edl muscle. Analyst 2010, 135, 3110–3119. [Google Scholar] [CrossRef] [PubMed]
- Fanning, K.M.; Pfisterer, B.; Davis, A.T.; Presley, T.D.; Williams, I.M.; Wasserman, D.H.; Cline, J.M.; Kavanagh, K. Changes in microvascular density differentiate metabolic health outcomes in monkeys with prior radiation exposure and subsequent skeletal muscle ECM remodeling. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R290–R297. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Lantier, L.; Kennedy, A.; Bonner, J.S.; Mayes, W.H.; Bracy, D.P.; Bookbinder, L.H.; Hasty, A.H.; Thompson, C.B.; Wasserman, D.H. Hyaluronan accumulates with high-fat feeding and contributes to insulin resistance. Diabetes 2013, 62, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Groen, B.B.; Hamer, H.M.; Snijders, T.; van Kranenburg, J.; Frijns, D.; Vink, H.; van Loon, L.J. Skeletal muscle capillary density and microvascular function are compromised with aging and type 2 diabetes. J. Appl. Physiol. 2014, 116, 998–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenmakers, A.J.; Strauss, J.A.; Shepherd, S.O.; Keske, M.A.; Cocks, M. Increased muscle blood supply and transendothelial nutrient and insulin transport induced by food intake and exercise: Effect of obesity and ageing. J. Physiol. 2016, 594, 2207–2222. [Google Scholar] [CrossRef] [PubMed]
- Prior, S.J.; Goldberg, A.P.; Ortmeyer, H.K.; Chin, E.R.; Chen, D.; Blumenthal, J.B.; Ryan, A.S. Increased skeletal muscle capillarization independently enhances insulin sensitivity in older adults after exercise training and detraining. Diabetes 2015, 64, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.R.; Machado, M.; de Jesus, N.; Gomes, F.; Lessa, M.A.; Bonomo, I.T.; Tibirica, E. Structural and functional microvascular alterations in a rat model of metabolic syndrome induced by a high-fat diet. Obesity 2013, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.K.; Kim, Y.W.; Choi, S.J.; Kim, J.Y.; Jeune, K.H.; Won, K.C.; Kim, J.K.; Koh, G.Y.; Park, S.Y. COMP-angiopoietin-1 enhances skeletal muscle blood flow and insulin sensitivity in mice. Am. J. Physiol. Endocrinol. Met. 2009, 297, E402–E409. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W., 3rd; Kang, L.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Ayala, J.E.; Bracy, D.P.; Julien, B.M.; Rottman, J.N.; Fueger, P.T.; Wasserman, D.H. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007, 56, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- El Annabi, S.; Gautier, N.; Baron, V. Focal adhesion kinase and src mediate integrin regulation of insulin receptor phosphorylation. FEBS Lett. 2001, 507, 247–252. [Google Scholar] [CrossRef]
- Bisht, B.; Goel, H.; Dey, C. Focal adhesion kinase regulates insulin resistance in skeletal muscle. Diabetologia 2007, 50, 1058–1069. [Google Scholar] [CrossRef] [PubMed]
- Bisht, B.; Srinivasan, K.; Dey, C.S. In vivo inhibition of focal adhesion kinase causes insulin resistance. J. Physiol. 2008, 586, 3825–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Mokshagundam, S.; Reuter, B.; Lark, D.S.; Sneddon, C.C.; Hennayake, C.; Williams, A.S.; Bracy, D.P.; James, F.D.; Pozzi, A. Integrin-linked kinase in muscle is necessary for the development of insulin resistance in diet-induced obese mice. Diabetes 2016, 65, 1590–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehti, T.M.; Silvennoinen, M.; Kivela, R.; Kainulainen, H.; Komulainen, J. Effects of streptozotocin-induced diabetes and physical training on gene expression of extracellular matrix proteins in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Met. 2006, 290, E900–E907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashraf, J.M.; Ansari, M.A.; Khan, H.M.; Alzohairy, M.A.; Choi, I. Green synthesis of silver nanoparticles and characterization of their inhibitory effects on ages formation using biophysical techniques. Sci. Rep. 2016, 6, 20414. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gautieri, A.; Passini, F.S.; Silvan, U.; Guizar-Sicairos, M.; Carimati, G.; Volpi, P.; Moretti, M.; Schoenhuber, H.; Redaelli, A.; Berli, M.; et al. Advanced glycation end-products: Mechanics of aged collagen from molecule to tissue. Matrix. Biol. 2017, 59, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eble, J.A.; de Rezende, F.F. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid. Redox. Signal. 2014, 20, 1977–1993. [Google Scholar] [CrossRef] [PubMed]
- Bartling, B.; Desole, M.; Rohrbach, S.; Silber, R.E.; Simm, A. Age-associated changes of extracellular matrix collagen impair lung cancer cell migration. FASEB J. 2009, 23, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Tarsio, J.F.; Reger, L.A.; Furcht, L.T. Decreased interaction of fibronectin, type iv collagen, and heparin due to nonenzymic glycation. Implications for diabetes mellitus. Biochemistry 1987, 26, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Pastino, A.K.; Greco, T.M.; Mathias, R.A.; Cristea, I.M.; Schwarzbauer, J.E. Stimulatory effects of advanced glycation endproducts (ages) on fibronectin matrix assembly. Matrix. Biol. 2017, 59, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.C.; Shiu, S.W.; Wong, Y.; Tam, X. Serum advanced glycation end products (ages) are associated with insulin resistance. Diabetes Metab. Res. Rev. 2011, 27, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Wahab, Y.H.; O’Harte, F.P.; Ratcliff, H.; McClenaghan, N.H.; Barnett, C.R.; Flatt, P.R. Glycation of insulin in the islets of langerhans of normal and diabetic animals. Diabetes 1996, 45, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Baig, M.H.; Jan, A.T.; Rabbani, G.; Ahmad, K.; Ashraf, J.M.; Kim, T.; Min, H.S.; Lee, Y.H.; Cho, W.-K.; Ma, J.Y. Methylglyoxal and advanced glycation end products: Insight of the regulatory machinery affecting the myogenic program and of its modulation by natural compounds. Sci. Rep. 2017, 7, 5916. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.; McAinch, A.J.; Dixon, J.B.; O’Brien, P.E.; Cameron-Smith, D. Increased Smad signaling and reduced MRF expression in skeletal muscle from obese subjects. Obesity 2013, 21, 525–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hittel, D.S.; Berggren, J.R.; Shearer, J.; Boyle, K.; Houmard, J.A. Increased secretion and expression of myostatin in skeletal muscle from extremely obese women. Diabetes 2009, 58, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Potes, Y.; de Luxán-Delgado, B.; Rodriguez-González, S.; Guimarães, M.R.M.; Solano, J.J.; Fernández-Fernández, M.; Bermúdez, M.; Boga, J.A.; Vega-Naredo, I.; Coto-Montes, A. Overweight in elderly people induces impaired autophagy in skeletal muscle. Free Radic. Biol. Med. 2017, 110, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.; Caldow, M.K.; Watts, R.; Levinger, P.; Cameron-Smith, D.; Levinger, I. Age and sex differences in human skeletal muscle fibrosis markers and transforming growth factor-β signaling. Eur. J. Appl. Physiol. 2017, 117, 1463–1472. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Bandinelli, S.; Sun, K.; Guralnik, J.M.; Ferrucci, L. Relationship of an advanced glycation end product, plasma carboxymethyl-lysine, with slow walking speed in older adults: The inchianti study. Eur. J. Appl. Physiol. 2010, 108, 191. [Google Scholar] [CrossRef] [PubMed]
- Dalal, M.; Ferrucci, L.; Sun, K.; Beck, J.; Fried, L.P.; Semba, R.D. Elevated serum advanced glycation end products and poor grip strength in older community-dwelling women. J. Gerontol. Ser. A: Biomed. Sci. Med. Sci. 2009, 64, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Momma, H.; Niu, K.; Kobayashi, Y.; Guan, L.; Sato, M.; Guo, H.; Chujo, M.; Otomo, A.; Yufei, C.; Tadaura, H. Skin advanced glycation end product accumulation and muscle strength among adult men. Eur. J. Appl. Physiol. 2011, 111, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Kubo, A.; Sugioka, Y.; Mitsui, R.; Fukuhara, N.; Nihei, F.; Takeda, Y. Relationship between advanced glycation end-product accumulation and low skeletal muscle mass in japanese men and women. Geriatr. Gerontol. Int. 2017, 17, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kuroda, A.; Araki, M.; Suzuki, R.; Taniguchi, S.; Tamaki, M.; Akehi, Y.; Matsuhisa, M. Advanced glycation end-products are a risk for muscle weakness in japanese patients with type 1 diabetes. J. Diabetes Investig. 2017, 8, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Jansson, E.; Fischer, H.; Gustafsson, T.; Greenhaff, P.L.; Ridden, J.; Rachman, J.; Sundberg, C.J. Modulation of extracellular matrix genes reflects the magnitude of physiological adaptation to aerobic exercise training in humans. BMC Biol. 2005, 3, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjorth, M.; Norheim, F.; Meen, A.J.; Pourteymour, S.; Lee, S.; Holen, T.; Jensen, J.; Birkeland, K.I.; Martinov, V.N.; Langleite, T.M. The effect of acute and long-term physical activity on extracellular matrix and serglycin in human skeletal muscle. Physiol. Rep. 2015, 3, 12473. [Google Scholar] [CrossRef] [PubMed]
- Hjorth, M.; Pourteymour, S.; Görgens, S.; Langleite, T.; Lee, S.; Holen, T.; Gulseth, H.; Birkeland, K.; Jensen, J.; Drevon, C. Myostatin in relation to physical activity and dysglycaemia and its effect on energy metabolism in human skeletal muscle cells. Acta Physiol. 2016, 217, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Nat. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garito, T.; Roubenoff, R.; Hompesch, M.; Morrow, L.; Gomez, K.; Rooks, D.; Meyers, C.; Buchsbaum, M.; Neelakantham, S.; Swan, T. Bimagrumab improves body composition and insulin sensitivity in insulin-resistant subjects. Diabetes Obes. Metab. 2017, 20, 94–102. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Collagen Types | Description | Expression During Diet-Induced IR | Reference |
|---|---|---|---|
| I | Abundantly found in endo-, peri-, and epimysium. Stimulate myogenic differentiation of stem cells. | ↑ | [16,33] |
| III | It is more consistently found between endomysium and epimysium. | ↑ | [34] |
| IV | Main component of basal lamina. Found to be 4 to 30-fold increase in skeletal muscle ECM mRNA levels | ↑ | [35] |
| V | Fibril-forming collagen and found to be increased in skeletal muscle ECM mRNA levels | [35] | |
| VI | Found to be increased in skeletal muscle ECM mRNA levels | [35] | |
| IX | Multiple-epiphyseal-dysplasia-related myopathy is caused due to mutation in collagen IX | [36] | |
| XII | It is the largest member of the fibril-associated collagens with interrupted triple helix (FACIT) family. Important for muscle integrity. | [37] | |
| XIV | A member of FACIT family and involved in muscle metabolism | [38] | |
| XV | Extensively found in the basement membrane and a structural component vital to stabilizing the skeletal muscle | [39] | |
| XVIII | Classified as multiplexins, bind with growth factors and other membranes of basement membrane glycoproteins. | [39] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, K.; Lee, E.J.; Moon, J.S.; Park, S.-Y.; Choi, I. Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle. Cells 2018, 7, 148. https://doi.org/10.3390/cells7100148
Ahmad K, Lee EJ, Moon JS, Park S-Y, Choi I. Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle. Cells. 2018; 7(10):148. https://doi.org/10.3390/cells7100148
Chicago/Turabian StyleAhmad, Khurshid, Eun Ju Lee, Jun Sung Moon, So-Young Park, and Inho Choi. 2018. "Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle" Cells 7, no. 10: 148. https://doi.org/10.3390/cells7100148
APA StyleAhmad, K., Lee, E. J., Moon, J. S., Park, S. -Y., & Choi, I. (2018). Multifaceted Interweaving Between Extracellular Matrix, Insulin Resistance, and Skeletal Muscle. Cells, 7(10), 148. https://doi.org/10.3390/cells7100148