Autophagy in Metabolic Age-Related Human Diseases



{kind=link}
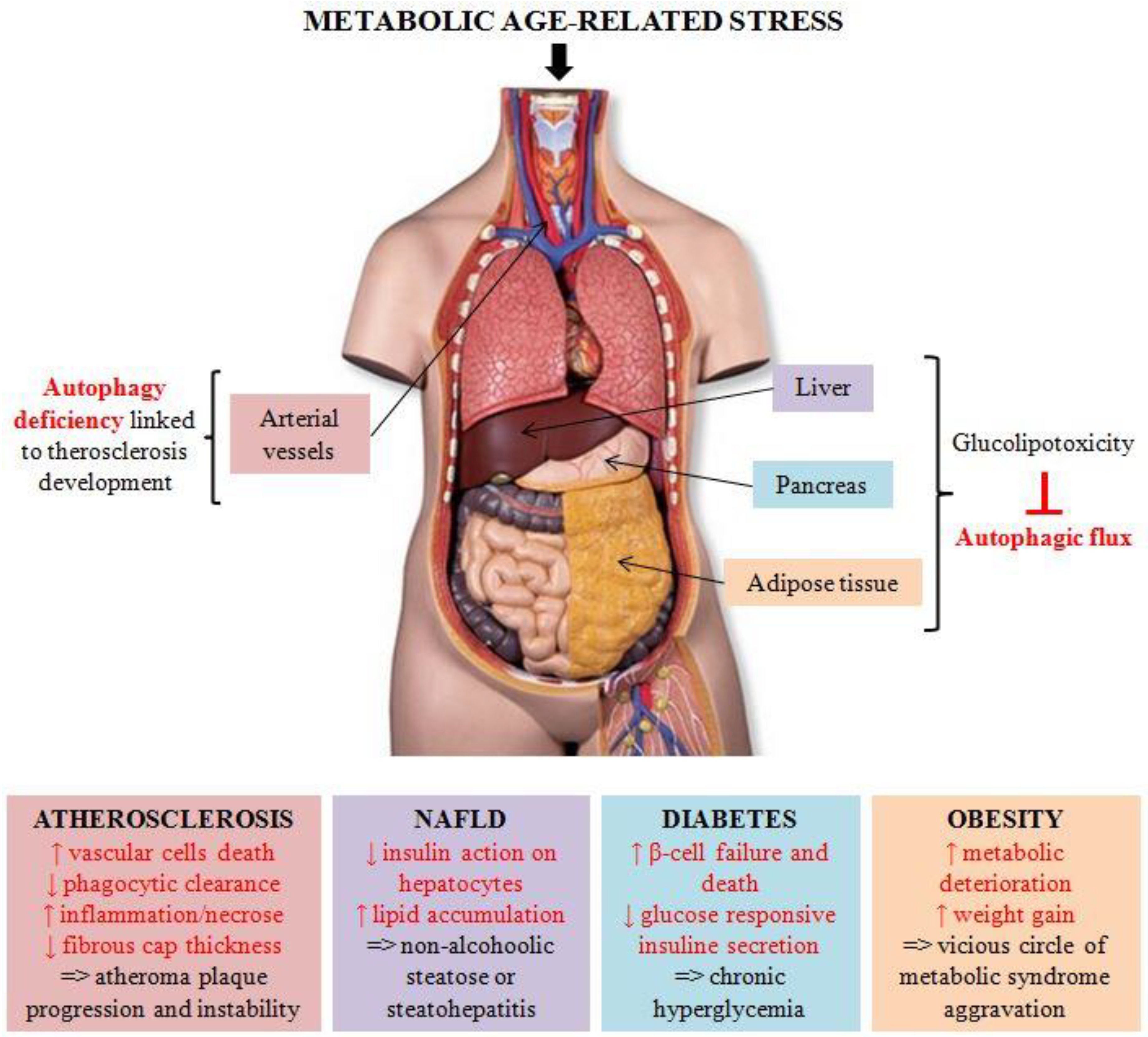
Abstract
:1. Introduction
2. Autophagy in Metabolic Diseases
2.1. Diabetes
2.1.1. Pancreatic β-Cell Function and Autophagy
2.1.2. Autophagy Involvement in Type 2 Diabetes Mellitus
2.2. Obesity
2.2.1. Nutrient Status and Autophagy
2.2.2. Dysregulated Autophagy in Obesity
2.3. Liver Diseases
2.3.1. Hepatic Metabolism and Autophagy
2.3.2. Hepatic Autophagy and Fatty Liver/Non-Alcoholic Steatohepatitis
2.4. Atherosclerosis
2.4.1. Vascular Function and Autophagy
2.4.2. Atherosclerosis and Dysregulated Autophagy
3. Pharmacological Modulation of Autophagy in Metabolic Age-Related Diseases
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Thumm, M.; Egner, R.; Koch, B.; Schlumpberger, M.; Straub, M.; Veenhuis, M.; Wolf, D.H. Isolation of autophagocytosis mutants of saccharomyces cerevisiae. FEBS Lett. 1994, 349, 275–280. [Google Scholar] [CrossRef]
- Harding, T.M.; Hefner-Gravink, A.; Thumm, M.; Klionsky, D.J. Genetic and phenotypic overlap between autophagy and the cytoplasm to vacuole protein targeting pathway. J. Biol Chem. 1996, 271, 17621–17624. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Moreau, K.; Jahreiss, L.; Puri, C.; Rubinsztein, D.C. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010, 12, 747–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 and Autophagy. Methods Mol. Biol. 2008, 445, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Ballabio, A. TFEB regulates autophagy: An integrated coordination of cellular degradation and recycling processes. Autophagy 2011, 7, 1379–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Watada, H.; Fujitani, Y. Minireview: Autophagy in pancreatic β-cells and its implication in diabetes. Mol. Endocrinol. Baltim. Md. 2015, 29, 338–348. [Google Scholar] [CrossRef] [PubMed]
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.-S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 2016, 59, 1480–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, H.L.; Peterson, B.S.; Haldeman, J.M.; Newgard, C.B.; Hohmeier, H.E.; Stephens, S.B. Delayed apoptosis allows islet β-cells to implement an autophagic mechanism to promote cell survival. PLoS ONE 2017, 12, e0172567. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won, K.J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.-H.; Kim, J.-W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Blandino-Rosano, M.; Barbaresso, R.; Jimenez-Palomares, M.; Bozadjieva, N.; Werneck-de-Castro, J.P.; Hatanaka, M.; Mirmira, R.G.; Sonenberg, N.; Liu, M.; Rüegg, M.A.; et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat. Commun. 2017, 8, 16014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.-I.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [PubMed]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.-J.; Wu, J.-H.; Sun, S.-Y.; Zhou, J.-Q. The endoplasmic reticulum stress/autophagy pathway is involved in cholesterol-induced pancreatic β-cell injury. Sci. Rep. 2017, 7, 44746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, R.P. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem. 2004, 279, 42351–42354. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fukutomi, T.; Sugawara, A.; Kawamura, H.; Takahashi, T.; Pi, J.; Uruno, A.; Yamamoto, M. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes 2014, 63, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.; Jiménez-Moreno, N.; García-Yagüe, Á.J.; Escoll, M.; de Ceballos, M.L.; Van Leuven, F.; Rábano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.-S.; Kim, J.; Lee, S.-J.; Kim, K.; Go, M.J.; Lee, J.-Y.; Lee, H.-J.; Song, J.; Jeon, B.T.; Roh, G.S.; et al. The PARK2 gene is involved in the maintenance of pancreatic β-cell functions related to insulin production and secretion. Mol. Cell. Endocrinol. 2014, 382, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, C.; Gao, J.; Xie, Z.; Chan, L.W.C.; Keating, D.J.; Yang, Y.; Sun, J.; Zhou, F.; Wei, Y.; et al. Inhibition of Miro1 disturbs mitophagy and pancreatic β-cell function interfering insulin release via IRS-Akt-Foxo1 in diabetes. Oncotarget 2017, 8, 90693–90705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goginashvili, A.; Zhang, Z.; Erbs, E.; Spiegelhalter, C.; Kessler, P.; Mihlan, M.; Pasquier, A.; Krupina, K.; Schieber, N.; Cinque, L.; et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic β cells. Science 2015, 347, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Marasco, M.R.; Linnemann, A.K. β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127–2141. [Google Scholar] [CrossRef] [PubMed]
- Martino, L.; Masini, M.; Novelli, M.; Beffy, P.; Bugliani, M.; Marselli, L.; Masiello, P.; Marchetti, P.; De Tata, V. Palmitate activates autophagy in INS-1E β-cells and in isolated rat and human pancreatic islets. PLoS ONE 2012, 7, e36188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Kong, F.; Pan, Q.; Du, Y.; Ye, J.; Zheng, F.; Li, H.; Zhou, J. Autophagy protects against cholesterol-induced apoptosis in pancreatic β-cells. Biochem. Biophys. Res. Commun. 2017, 482, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.M.; Bak, D.H.; Kim, D.H.; Hong, J.Y.; Han, S.Y.; Park, K.Y.; Lim, K.; Lim, D.M.; Kang, J.G. Omega-3 polyunsaturated fatty acids may attenuate streptozotocin-induced pancreatic β-cell death via autophagy activation in fat1 transgenic mice. Endocrinol. Metab. (Seoul) 2015, 30, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, D.; Ni, C.; Zhou, H.; Wu, S.; Xue, Z.; Zhou, Z. Vitamin D induces autophagy of pancreatic β-cells and enhances insulin secretion. Mol. Med. Rep. 2016, 14, 2644–2650. [Google Scholar] [CrossRef] [PubMed]
- Zummo, F.P.; Cullen, K.S.; Honkanen-Scott, M.; Shaw, J.A.M.; Lovat, P.E.; Arden, C. Glucagon-like peptide 1 protects pancreatic β-cells from death by increasing autophagic flux and restoring lysosomal function. Diabetes 2017, 66, 1272–1285. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, J.; Wu, H.; Liu, X.; Chen, Y.; Wu, J.; Hu, C.; Zou, D. Liraglutide protects pancreatic β-cells against free fatty acids in vitro and affects glucolipid metabolism in apolipoprotein E-/- mice by activating autophagy. Mol. Med. Rep. 2015, 12, 4210–4218. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.W.; Jin, L.; Jin, J.; Yang, C.W. Effect of exendin-4 on autophagy clearance in beta cell of rats with tacrolimus-induced diabetes mellitus. Sci. Rep. 2016, 6, 29921. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, J.; Yu, X. Dipeptidyl peptidase-4 inhibitor MK-626 restores insulin secretion through enhancing autophagy in high fat diet-induced mice. Biochem. Biophys. Res. Commun. 2016, 470, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Cheon, H.; Jeong, Y.T.; Quan, W.; Kim, K.H.; Cho, J.M.; Lim, Y.-M.; Oh, S.H.; Jin, S.-M.; Kim, J.H.; et al. Amyloidogenic peptide oligomer accumulation in autophagy-deficient β cells induces diabetes. J. Clin. Investig. 2014, 124, 3311–3324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.-S.; Lu, C.-C.; Kuo, S.-C.; Hsu, Y.-M.; Tsai, S.-C.; Chen, S.-Y.; Chen, Y.-T.; Lin, Y.-J.; Huang, Y.-C.; Chen, C.-J.; et al. Autophagy and its link to type II diabetes mellitus. BioMedicine 2017, 7, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, S.U.R.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of autophagic turnover in β-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.M.; Colby, A.H.; Zeng, J.; Las, G.; Feng, J.H.; Grinstaff, M.W.; Shirihai, O.S. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J. Cell Biol. 2016, 214, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, Y.-M.; Lim, H.; Hur, K.Y.; Quan, W.; Lee, H.-Y.; Cheon, H.; Ryu, D.; Koo, S.-H.; Kim, H.L.; Kim, J.; et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat. Commun. 2014, 5, 4934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, W.; Hur, K.Y.; Lim, Y.; Oh, S.H.; Lee, J.-C.; Kim, K.H.; Kim, G.H.; Kim, S.-W.; Kim, H.L.; Lee, M.-K.; et al. Autophagy deficiency in beta cells leads to compromised unfolded protein response and progression from obesity to diabetes in mice. Diabetologia 2012, 55, 392–403. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Xiao, X.; Prasadan, K.; Chen, C.; Ming, Y.; Fusco, J.; Gangopadhyay, N.N.; Ricks, D.; Gittes, G.K. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci. Rep. 2017, 7, 16348. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.Y.; O’Reilly, L.; Ramm, G.; Biden, T.J. High-fat diet increases autophagic flux in pancreatic beta cells in vivo and ex vivo in mice. Diabetologia 2015, 58, 2074–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.-O.; Yoo, S.-M.; Ahn, H.-H.; Nah, J.; Hong, S.-H.; Kam, T.-I.; Jung, S.; Jung, Y.-K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Shigihara, N.; Fukunaka, A.; Hara, A.; Komiya, K.; Honda, A.; Uchida, T.; Abe, H.; Toyofuku, Y.; Tamaki, M.; Ogihara, T.; et al. Human IAPP-induced pancreatic β cell toxicity and its regulation by autophagy. J. Clin. Investig. 2014, 124, 3634–3644. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.F.; Costes, S.; Gurlo, T.; Glabe, C.G.; Butler, P.C. Autophagy defends pancreatic β cells from human islet amyloid polypeptide-induced toxicity. J. Clin. Investig. 2014, 124, 3489–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-H.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet amyloid polypeptide: Structure, function, and pathophysiology. J. Diabetes Res. 2016, 2016, 2798269. [Google Scholar] [CrossRef] [PubMed]
- Bröer, S.; Bröer, A. Amino acid homeostasis and signalling in mammalian cells and organisms. Biochem. J. 2017, 474, 1935–1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Lee, M.-S. Autophagy—A key player in cellular and body metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell. 2011, 44, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Guo, S.; Gao, J.; Zhong, M.; Yan, G.; Wu, W.; Chao, Y.; Jiang, Y. General Control Nonderepressible 2 (GCN2) Kinase Inhibits Target of Rapamycin Complex 1 in Response to Amino Acid Starvation in Saccharomyces cerevisiae. J. Biol. Chem. 2017, 292, 2660–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sowers, J.R.; Ren, J. Targeting autophagy in obesity: From pathophysiology to management. Nat. Rev. Endocrinol. 2018, 14, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell. Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. Off Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Komiya, K.; Uchida, T.; Ueno, T.; Koike, M.; Abe, H.; Hirose, T.; Kawamori, R.; Uchiyama, Y.; Kominami, E.; Fujitani, Y.; et al. Free fatty acids stimulate autophagy in pancreatic β-cells via JNK pathway. Biochem. Biophys. Res. Commun. 2010, 401, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.; Bay, B.-H.; Wenk, M.R.; Shen, H.-M. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.B.; Olzmann, J.A. Lipid droplets and lipotoxicity during autophagy. Autophagy 2017, 13, 2002–2003. [Google Scholar] [CrossRef] [PubMed]
- Maixner, N.; Kovsan, J.; Harman-Boehm, I.; Blüher, M.; Bashan, N.; Rudich, A. Autophagy in adipose tissue. Obes. Facts 2012, 5, 710–721. [Google Scholar] [CrossRef] [PubMed]
- Maixner, N.; Bechor, S.; Vershinin, Z.; Pecht, T.; Goldstein, N.; Haim, Y.; Rudich, A. Transcriptional Dysregulation of Adipose Tissue Autophagy in Obesity. Physiol. Bethesda Md. 2016, 31, 270–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuñez, C.E.; Rodrigues, V.S.; Gomes, F.S.; Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Longo, V.D.; Harvie, M. Impact of intermittent fasting on health and disease processes. Ageing Res. Rev. 2017, 39, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Van Niekerk, G.; du Toit, A.; Loos, B.; Engelbrecht, A.-M. Nutrient excess and autophagic deficiency: Explaining metabolic diseases in obesity. Metabolism 2018, 82, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Zylke, J.W.; Bauchner, H. The Unrelenting Challenge of Obesity. JAMA 2016, 315, 2277–2278. [Google Scholar] [CrossRef] [PubMed]
- Namkoong, S.; Cho, C.-S.; Semple, I.; Lee, J.H. Autophagy dysregulation and obesity-associated pathologies. Mol. Cells 2018, 41, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Dann, S.G.; Selvaraj, A.; Thomas, G. mTOR Complex1-S6K1 signaling: At the crossroads of obesity, diabetes and cancer. Trends Mol. Med. 2007, 13, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.B.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed]
- Ost, A.; Svensson, K.; Ruishalme, I.; Brännmark, C.; Franck, N.; Krook, H.; Sandström, P.; Kjolhede, P.; Strålfors, P. Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. Camb. Mass. 2010, 16, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kosacka, J.; Kern, M.; Klöting, N.; Paeschke, S.; Rudich, A.; Haim, Y.; Gericke, M.; Serke, H.; Stumvoll, M.; Bechmann, I.; et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol. Cell. Endocrinol. 2015, 409, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Yan, W.-J.; Yin, T.-L.; Zhang, Y.; Li, J.; Yang, J. Diet-induced obesity impairs spermatogenesis: A potential role for autophagy. Sci. Rep. 2017, 7, 43475. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Kim, J.; Quan, W.; Lee, J.-C.; Kim, M.-S.; Kim, S.-H.; Bae, J.-W.; Hur, K.Y.; Lee, M.-S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Kim, H.-K.; Moon, E.-Y.; Kim, S.S.; Choi, C.S.; Komatsu, M.; Jeong, Y.T.; Lee, M.-K.; Kim, K.-W.; Kim, M.-S.; et al. Role of hypothalamic proopiomelanocortin neuron autophagy in the control of appetite and leptin response. Endocrinology 2012, 153, 1817–1826. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-J.R.; Kim, H.; Oh, S.; Lee, J.-G.; Kee, M.; Ko, H.-J.; Kweon, M.-N.; Won, K.-J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Turdi, S.; Richmond, K.L.; Zhang, Y.; Ren, J. ALDH2 protects against high fat diet-induced obesity cardiomyopathy and defective autophagy: Role of CaM kinase II.; histone H3K9 methyltransferase SUV39H.; Sirt1.; and PGC-1α deacetylation. Int. J. Obes. 2018, 42, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Ashford, T.P.; Porter, K.R. Cytoplasmic components in hepatic cell lysosomes. J. Cell Biol. 1962, 12, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Schworer, C.M.; Shiffer, K.A.; Mortimore, G.E. Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J. Biol. Chem. 1981, 256, 7652–7658. [Google Scholar] [PubMed]
- Mortimore, G.E.; Hutson, N.J.; Surmacz, C.A. Quantitative correlation between proteolysis and macro- and microautophagy in mouse hepatocytes during starvation and refeeding. Proc. Natl. Acad. Sci. USA 1983, 80, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- Schworer, C.M.; Mortimore, G.E. Glucagon-induced autophagy and proteolysis in rat liver: Mediation by selective deprivation of intracellular amino acids. Proc. Natl. Acad. Sci. USA 1979, 76, 3169–3173. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Fang, W.; Chen, X.; Lin, Y.; Hu, G.; Wei, J.; Zhang, X.; Yang, C.; Li, J. Upstream stimulating factor 1 suppresses autophagy and hepatic lipid droplet catabolism by activating mTOR. FEBS Lett. 2018, 592, 2725–2738. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat. Cell Biol. 2015, 17, 759–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, G.; Han, D.H.; Lee, M.; Kim, I.; Kim, B.; Kim, K.H.; Song, Y.M.; Yoo, J.E.; Wang, H.J.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Chang, P.F.; Lin, H.F.; Liu, K.; Chang, M.H.; Ni, Y.H. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J. Hepatol. 2016, 65, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Torisu, T.; Torisu, K.; Lee, I.H.; Liu, J.; Malide, D.; Combs, C.A.; Wu, X.S.; Rovira, I.; Fergusson, M.M.; Weigert, R.; et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat. Med. 2013, 19, 1281–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Li, X.; Peng, J.; Tang, Y.; Yang, Q.; Liu, L.; Wang, Z.; Jiang, Z.; Xiao, M.; Ni, C.; et al. Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann. Biomed. Eng. 2014, 42, 1978–1988. [Google Scholar] [CrossRef] [PubMed]
- Yao, P.; Zhao, H.; Mo, W.; He, P. Laminar Shear Stress Promotes Vascular Endothelial Cell Autophagy Through Upregulation with Rab4. DNA Cell Biol. 2016, 35, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bi, X.; Chen, T.; Zhang, Q.; Wang, S.-X.; Chiu, J.-J.; Liu, G.-S.; Zhang, Y.; Bu, P.; Jiang, F. Shear stress regulates endothelial cell autophagy via redox regulation and Sirt1 expression. Cell Death Dis. 2015, 6, e1827. [Google Scholar] [CrossRef] [PubMed]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 114, E8675–E8684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Teng, R.J.; Guan, T.; Eis, A.; Kaul, S.; Konduri, G.G.; Shi, Y. Role of autophagy in angiogenesis in aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 302, C383–C391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salabei, J.K.; Cummins, T.D.; Singh, M.; Jones, S.P.; Bhatnagar, A.; Hill, B.G. PDGF-mediated autophagy regulates vascular smooth muscle cell phenotype and resistance to oxidative stress. Biochem. J. 2013, 451, 375–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C.F.; Fransen, P.; De Munck, D.G.; De Meyer, G.R.; Martinet, W. Defective autophagy in vascular smooth muscle cells alters contractility and Ca2+ homeostasis in mice. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H557–H567. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Y.; Zhao, M.-M.; Cai, Y.; Guan, Q.-C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.-G.; Xu, M.-J.; Wang, X. Phosphate-induced autophagy counteracts vascular calcification by reducing matrix vesicle release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Wang, X.; Schnackenberg, L.; Khaidakov, M.; Liu, S.; Singla, S.; Dai, Y.; Mehta, J.L. Regulation of autophagy and apoptosis in response to ox-LDL in vascular smooth muscle cells, and the modulatory effects of the microRNA hsa-let-7 g. Int. J. Cardiol. 2013, 168, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Yang, Y.; Yan, M.; Zhan, J.; Fu, X.; Zheng, X. Autophagy plays a protective role in free cholesterol overload-induced death of smooth muscle cells. J. Lipid Res. 2010, 51, 2581–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Marcel, Y.L. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Vindis, C. Autophagy: An emerging therapeutic target in vascular diseases. Br. J. Pharmacol. 2015, 172, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, G.R.Y.; Grootaert, M.O.J.; Michiels, C.F.; Kurdi, A.; Schrijvers, D.M.; Martinet, W. Autophagy in vascular disease. Circ. Res. 2015, 116, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Salvayre, R.; Negre-Salvayre, A.; Vindis, C. HDLs inhibit endoplasmic reticulum stress and autophagic response induced by oxidized LDLs. Cell Death Differ. 2011, 18, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Salvayre, R.; Nègre-Salvayre, A.; Vindis, C. Oxidized LDLs trigger endoplasmic reticulum stress and autophagy: Prevention by HDLs. Autophagy 2011, 7, 541–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiader, A.; Nahapetyan, H.; Faccini, J.; D’Angelo, R.; Mucher, E.; Elbaz, M.; Boya, P.; Vindis, C. Mitophagy acts as a safeguard mechanism against human vascular smooth muscle cell apoptosis induced by atherogenic lipids. Oncotarget 2016, 7, 28821–28835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.M. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osonoi, Y.; Mita, T.; Azuma, K.; Nakajima, K.; Masuyama, A.; Goto, H.; Nishida, Y.; Miyatsuka, T.; Fujitani, Y.; Koike, M.; et al. Defective autophagy in vascular smooth muscle cells enhances cell death and atherosclerosis. Autophagy 2018, 14, 1991–2006. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Feng, C.; Coleman, T.; Emanuel, R.; Wen, H.; Hwang, S.; Ting, J.P.; Virgin, H.W.; Kastan, M.B.; Semenkovich, C.F. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15, 534–544. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Sergin, I.; Bhattacharya, S.; Emanuel, R.; Esen, E.; Stokes, C.J.; Evans, T.D.; Arif, B.; Curci, J.A.; Razani, B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci. Signal. 2016, 9, ra2. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Autophagy: A druggable process that is deregulated in aging and human disease. J. Clin. Investig. 2015, 125, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Sequea, D.A.; Sharma, N.; Arias, E.B.; Cartee, G.D. Calorie restriction enhances insulin-stimulated glucose uptake and Akt phosphorylation in both fast-twitch and slow-twitch skeletal muscle of 24-month-old rats. J. Gerontol. Biol. Sci. Med. Sci. 2012, 67, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Marcinko, K.; Sikkema, S.R.; Samaan, M.C.; Kemp, B.E.; Fullerton, M.D.; Steinberg, G.R. High intensity interval training improves liver and adipose tissue insulin sensitivity. Mol. Metab. 2015, 4, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Leontieva, O.V.; Paszkiewicz, G.; Demidenko, Z.N.; Blagosklonny, M.V. Resveratrol potentiates rapamycin to prevent hyperinsulinemia and obesity in male mice on high fat diet. Cell Death Dis. 2013, 4, e472. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, J.; Hou, J.; Lin, C.; Bensoussan, A.; Chang, D.; Liu, J.; Wang, B. Berberine alleviates ox-LDL induced inflammatory factors by up-regulation of autophagy via AMPK/mTOR signaling pathway. J. Transl. Med. 2015, 13, 92. [Google Scholar] [CrossRef] [PubMed]
- Tillhon, M.; Guamán Ortiz, L.M.; Lombardi, P.; Scovassi, A.I. Berberine: New perspectives for old remedies. Biochem. Pharmacol. 2012, 84, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [PubMed]
- Sadasivan, S.K.; Vasamsetti, B.; Singh, J.; Marikunte, V.V.; Oommen, A.M.; Jagannath, M.R.; Pralhada Rao, R. Exogenous administration of spermine improves glucose utilization and decreases bodyweight in mice. Eur. J. Pharmacol. 2014, 729, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Higgins, C.B.; Mayer, A.L.; Mysorekar, I.U.; Razani, B.; Graham, M.J.; Hruz, P.W.; DeBosch, B.J. TFEB-dependent induction of thermogenesis by the hepatocyte SLC2A inhibitor trehalose. Autophagy 2018, 14, 1959–1975. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Han, D.H.; Nam, K.T.; Park, J.S.; Kim, S.H.; Lee, M.; Kim, G.; Min, B.S.; Cha, B.S.; Lee, Y.S.; et al. Ezetimibe, an NPC1L1 inhibitor, is a potent Nrf2 activator that protects mice from diet-induced nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2016, 99, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Park, H.; Semple, I.A.; Jang, I.; Ro, S.H.; Kim, M.; Cazares, V.A.; Stuenkel, E.L.; Kim, J.J.; Kim, J.S.; et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat. Commun. 2014, 5, 4834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verheye, S.; Martinet, W.; Kockx, M.M.; Knaapen, M.W.; Salu, K.; Timmermans, J.P.; Ellis, J.T.; Kilpatrick, D.L.; De Meyer, G.R. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J. Am. Coll. Cardiol. 2007, 49, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Ye, L.; Katajisto, P.; Goncalves, M.D.; Saitoh, M.; Stevens, D.M.; Davis, J.G.; Salmon, A.B.; Richardson, A.; Ahima, R.S.; et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012, 335, 1638–1643. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moulis, M.; Vindis, C. Autophagy in Metabolic Age-Related Human Diseases. Cells 2018, 7, 149. https://doi.org/10.3390/cells7100149
Moulis M, Vindis C. Autophagy in Metabolic Age-Related Human Diseases. Cells. 2018; 7(10):149. https://doi.org/10.3390/cells7100149
Chicago/Turabian StyleMoulis, Manon, and Cecile Vindis. 2018. "Autophagy in Metabolic Age-Related Human Diseases" Cells 7, no. 10: 149. https://doi.org/10.3390/cells7100149
APA StyleMoulis, M., & Vindis, C. (2018). Autophagy in Metabolic Age-Related Human Diseases. Cells, 7(10), 149. https://doi.org/10.3390/cells7100149