The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
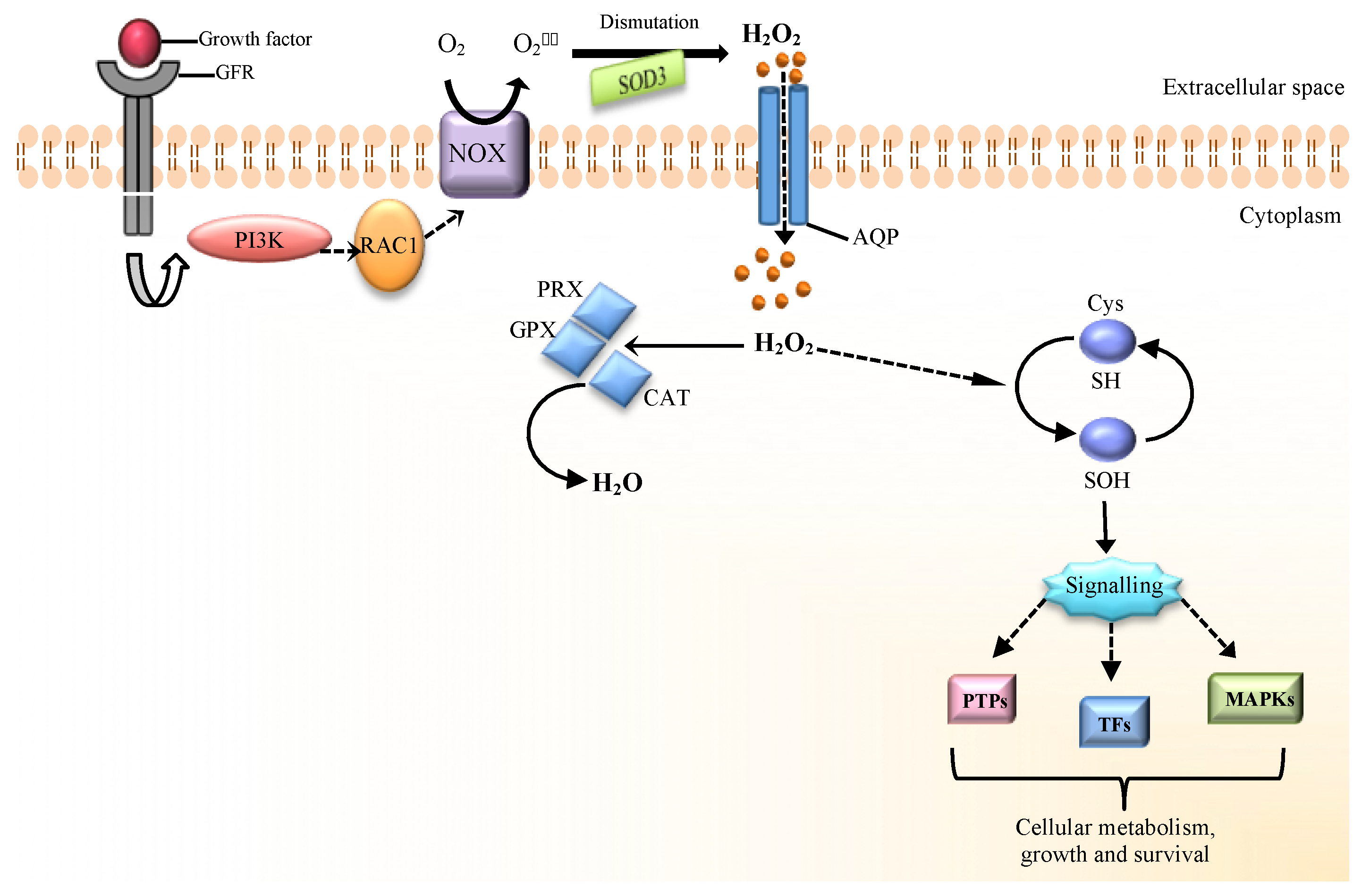
2. The Source of H2O2 at the Beginning of Redox Signaling
3. H2O2 as a Molecular Mediator of Cellular Signaling
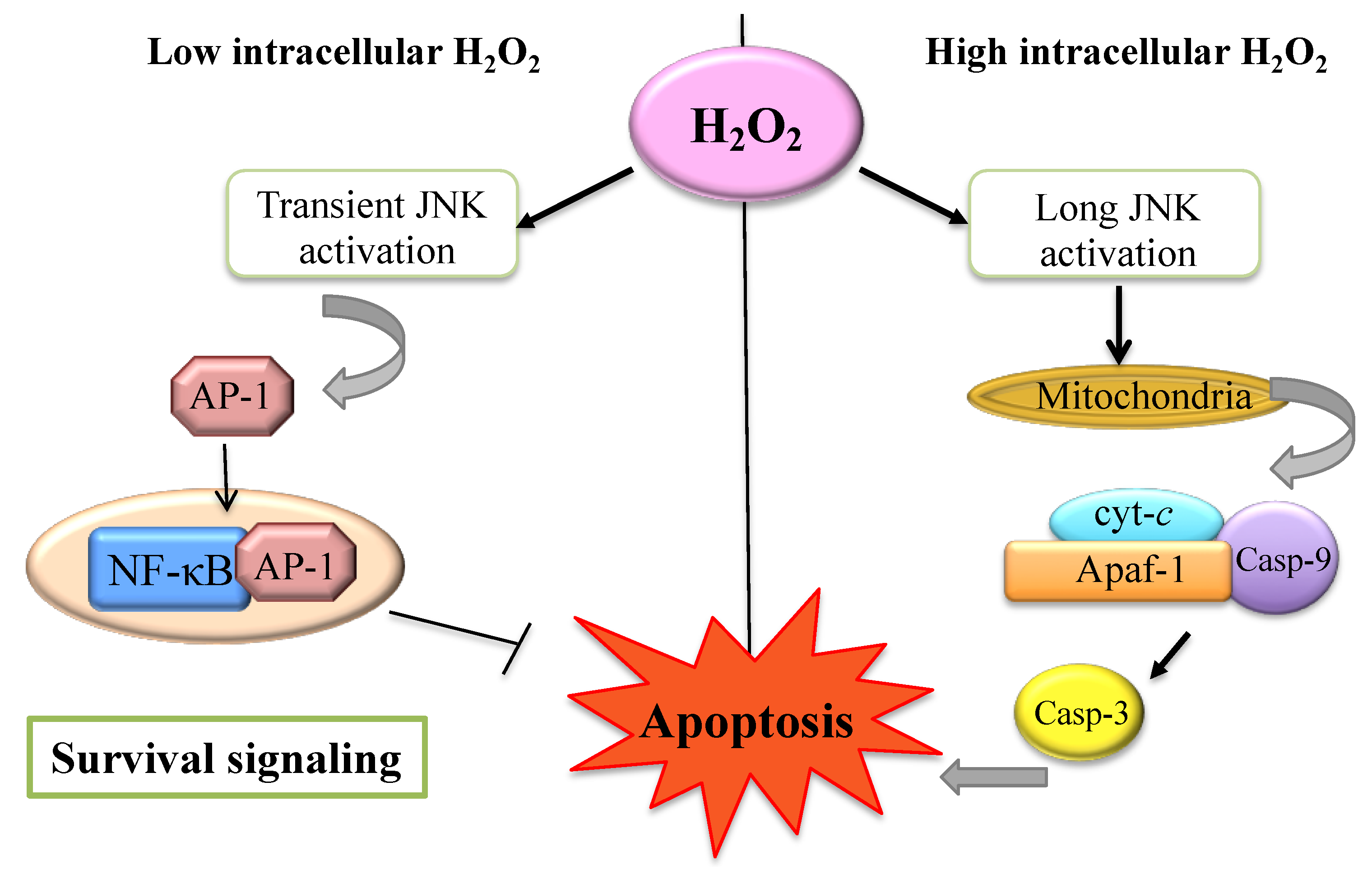
4. The H2O2 within the Cell: A Matter of Concentration
5. H2O2 and Apoptosis
6. H2O2, Bacteria, and Immune Cells
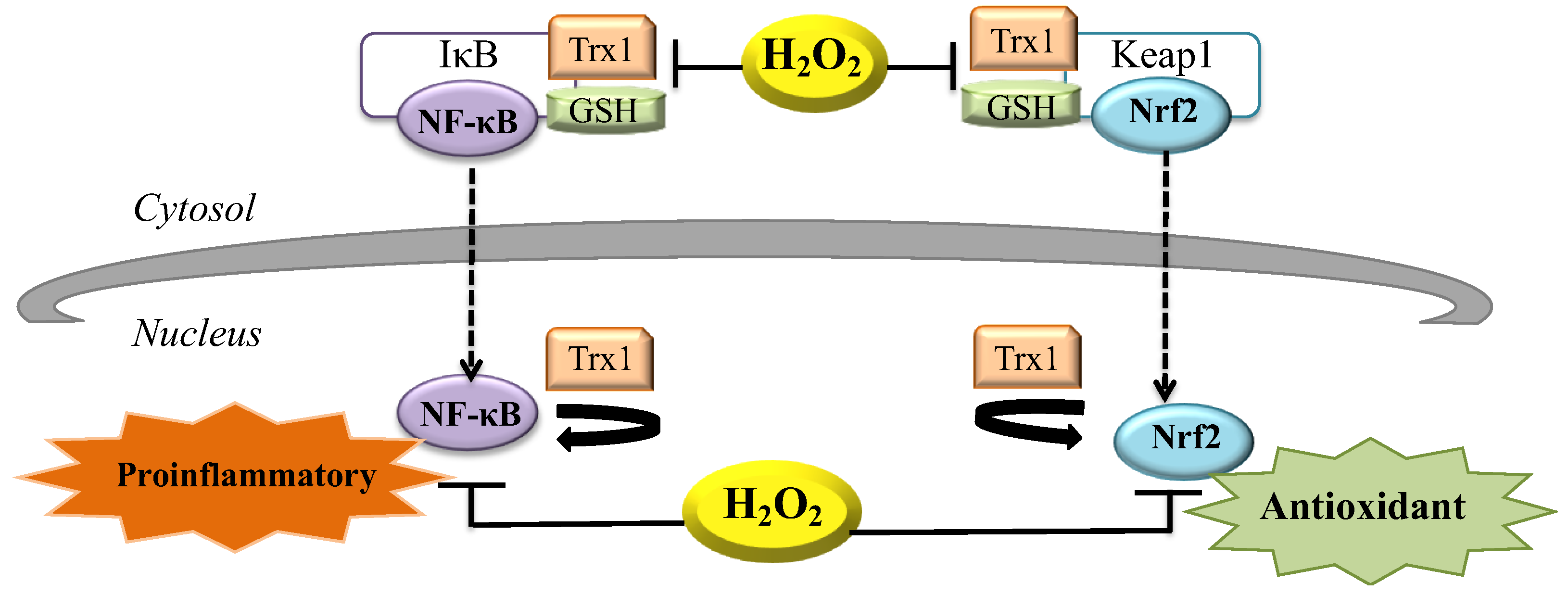
7. H2O2 during Inflammation
8. H2O2 and the Control of Gene Expression
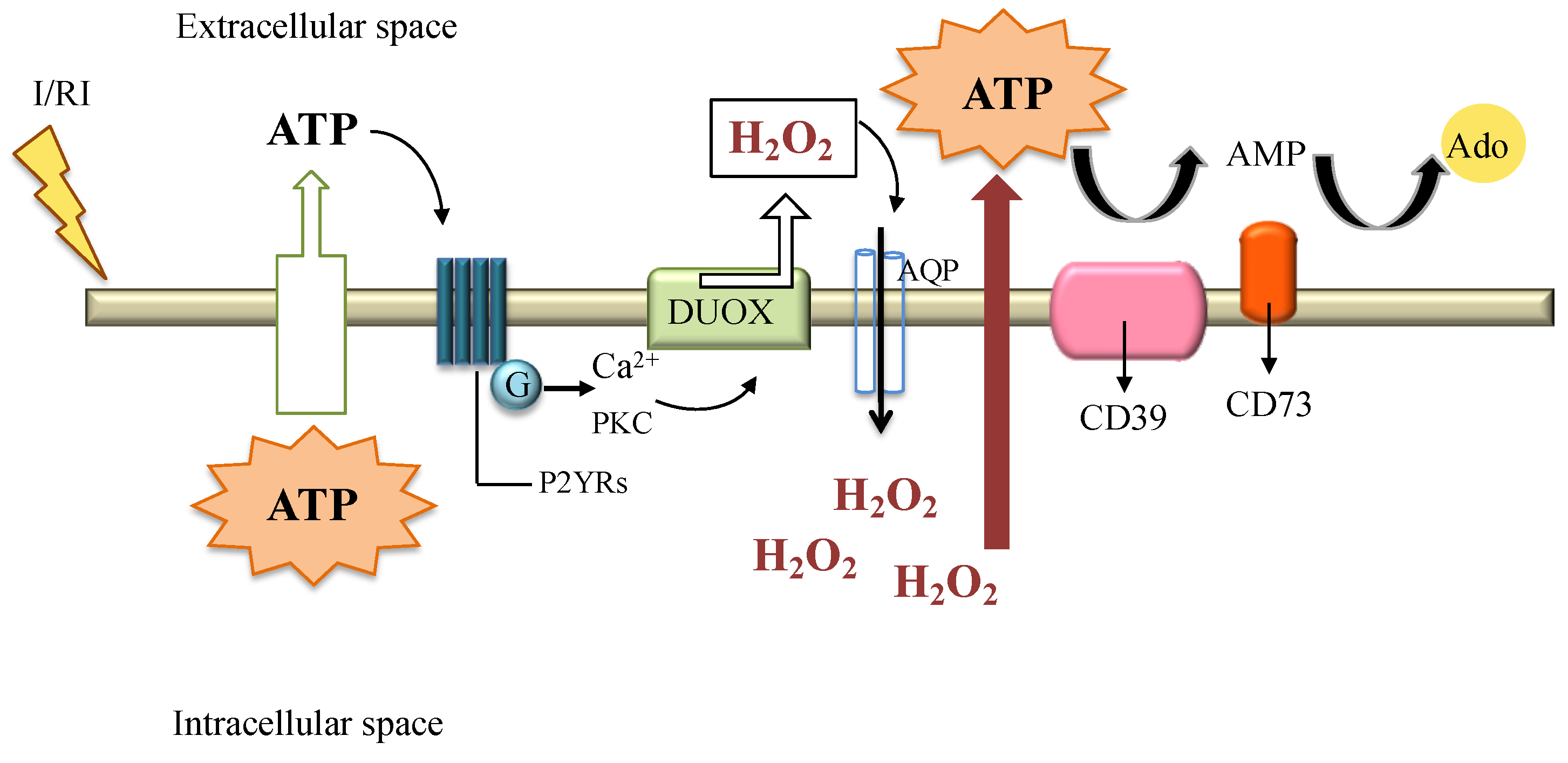
9. H2O2 in Disease Settings: Ischemia Reperfusion Injury
10. H2O2 and the Protective Signaling Pathways: The Ectonucleotidases
11. H2O2 and the Protective Signaling Pathways: The Heme Oxygenase-1
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Linley, E.; Denyer, S.P.; McDonnell, G.; Simons, C.; Maillard, J.-Y. Use of hydrogen peroxide as a biocide: New consideration of its mechanisms of biocidal action. J. Antimicrob. Chemother. 2012, 67, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.-M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid. Med. Cell. Longev. 2017, 2017, 8459402. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide- production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.-F. Superoxide dismutases: Ancient enzymes and new insights. FEBS Lett. 2012, 586, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Sumimoto, H.; Miyano, K.; Takeya, R. Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem. Biophys. Res. Commun. 2005, 338, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- Vieceli Dalla Sega, F.; Zambonin, L.; Fiorentini, D.; Rizzo, B.; Caliceti, C.; Landi, L.; Hrelia, S.; Prata, C. Specific aquaporins facilitate Nox-produced hydrogen peroxide transport through plasma membrane in leukaemia cells. Biochim. Biophys. Acta 2014, 1843, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.-Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.-C.; Fukada, T.; Tonks, N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.-H.; Lee, C.-H.; Ahn, Y.; Kim, H.; Kim, H.; Ahn, C.-Y.; Yang, K.-S.; Lee, S.-R. Redox regulation of PTEN and protein tyrosine phosphatases in H2O2 mediated cell signaling. FEBS Lett. 2004, 560, 7–13. [Google Scholar] [CrossRef]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef] [PubMed]
- Reczek, C.R.; Chandel, N.S. ROS-dependent signal transduction. Curr. Opin. Cell Biol. 2015, 33, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013, 1, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-J.; Wang, M.-J.; Moore, P.K.; Jin, H.-M.; Yao, T.; Zhu, Y.-C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Q.; Wang, B.; Zhang, X.-F.; Ma, Y.-P.; Liu, J.-D.; Wang, X.-Z. Contribution of hydrogen sulfide and nitric oxide to exercise-induced attenuation of aortic remodeling and improvement of endothelial function in spontaneously hypertensive rats. Mol. Cell. Biochem. 2012, 375, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.W.; Jha, S.; Gundewar, S.; Elrod, J.W.; Ramachandran, A.; Pattillo, C.B.; Kevil, C.G.; Lefer, D.J. Hydrogen Sulfide Mediates Cardioprotection Through Nrf2 Signaling. Circ. Res. 2009, 105, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-Y.; Chu, H.-M.; Pan, K.-T.; Teng, C.-H.; Wang, D.-L.; Wang, A.H.J.; Khoo, K.-H.; Meng, T.-C. Cysteine S-nitrosylation protects protein-tyrosine phosphatase 1B against oxidation-induced permanent inactivation. J. Biol. Chem. 2008, 283, 35265–35272. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 2012, 5, pe10. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, J.R. Protein cysteine thiol nitrosation: Maker or marker of reactive nitrogen species-induced nonerythroid cellular signaling? Nitric Oxide 2008, 19, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [PubMed]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.S.; Antunes, F. The Roles of Peroxiredoxin and Thioredoxin in Hydrogen Peroxide Sensing and in Signal Transduction. Mol. Cells 2016, 39, 65–71. [Google Scholar] [PubMed] [Green Version]
- Kanta, J. The role of hydrogen peroxide and other reactive oxygen species in wound healing. Acta Medica 2011, 54, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B. Investigating the role of reactive oxygen species in regulating autophagy. Methods Enzymol. 2013, 528, 217–235. [Google Scholar] [PubMed]
- Xiang, J.; Wan, C.; Guo, R.; Guo, D. Is Hydrogen Peroxide a Suitable Apoptosis Inducer for All Cell Types? Biomed. Res. Int. 2016, 2016, 7343965. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Vita, J.A.; Berk, B.C.; Keaney, J.F. c-Jun N-terminal kinase activation by hydrogen peroxide in endothelial cells involves SRC-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 2001, 276, 16045–16050. [Google Scholar] [CrossRef] [PubMed]
- Drane, P.; Bravard, A.; Bouvard, V.; May, E. Reciprocal down-regulation of p53 and SOD2 gene expression-implication in p53 mediated apoptosis. Oncogene 2001, 20, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Bedogni, B.; Anzevino, R.; Colavitti, R.; Palazzotti, B.; Borrello, S.; Galeotti, T. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res. 2000, 60, 4654–4660. [Google Scholar] [PubMed]
- Liu, D.; Xu, Y. p53, oxidative stress, and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; He, P.; Robles, A.; Lupold, S.; Kaneko, I.; Ichimiya, M.; Sengupta, S.; Mechanic, L.; Okamura, S.; et al. p53-Induced Up-Regulation of MnSOD and GPx but not Catalase Increases Oxidative Stress and Apoptosis. Cancer Res. 2004, 64, 2350–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Transcriptional regulation by MAP kinases. Mol. Reprod. Dev. 1995, 42, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Baas, A.S.; Berk, B.C. Differential activation of mitogen-activated protein kinases by H2O2 and O2- in vascular smooth muscle cells. Circ. Res. 1995, 77, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Guyton, K.Z.; Liu, Y.; Gorospe, M.; Xu, Q.; Holbrook, N.J. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J. Biol. Chem. 1996, 271, 4138–4142. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, C.; Chockley, P.; Singh, S.K.; Pase, L.; Lieschke, G.J.; Grabher, C. Hydrogen peroxide in inflammation: Messenger, guide, and assassin. Adv. Hematol. 2012, 2012, 541471. [Google Scholar] [CrossRef] [PubMed]
- Van der Vliet, A.; Janssen-Heininger, Y.M.W. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell. Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef] [PubMed]
- García-Redondo, A.B.; Aguado, A.; Briones, A.M.; Salaices, M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016, 114, 110–120. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A. Role of the NADPH oxidase systems Nox and Duox in host defense and inflammation. Expert Rev. Clin. Immunol. 2007, 3, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Chandler, J.D.; Day, B.J. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radic. Res. 2015, 49, 695–710. [Google Scholar] [CrossRef] [PubMed]
- Allaoui, A.; Botteaux, A.; Dumont, J.E.; Hoste, C.; De Deken, X. Dual oxidases and hydrogen peroxide in a complex dialogue between host mucosae and bacteria. Trends Mol. Med. 2009, 15, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belikov, A.V.; Schraven, B.; Simeoni, L. T cells and reactive oxygen species. J. Biomed. Sci. 2015, 22, 85. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, S.; Kabelitz, D. When neutrophils meet T cells: Beginnings of a tumultuous relationship with underappreciated potential. Eur. J. Immunol. 2014, 44, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Y.; Wang, K.; Deng, L.; Chen, Y.; Nice, E.C.; Huang, C. Redox regulation of inflammation: Old elements, a new story. Med. Res. Rev. 2015, 35, 306–340. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D. The Rel/NF-κB signal transduction pathway: Introduction. Oncogene 1999, 18, 6842–6844. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-κB is activated: The role of the IκB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Schreck, R.; Rieber, P.; Baeuerle, P.A. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-κB transcription factor and HIV-1. EMBO J. 1991, 10, 2247–2258. [Google Scholar] [CrossRef] [PubMed]
- Halvey, P.J.; Hansen, J.M.; Johnson, J.M.; Go, Y.-M.; Samali, A.; Jones, D.P. Selective oxidative stress in cell nuclei by nuclear-targeted D-amino acid oxidase. Antioxid. Redox Signal. 2007, 9, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Cebula, M.; Schmidt, E.E.; Arnér, E.S.J. TrxR1 as a Potent Regulator of the Nrf2-Keap1 Response System. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Carden, D.L.; Granger, D.N. Pathophysiology of ischaemia-reperfusion injury. J. Pathol. 2000, 190, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Grisham, M.B.; Granger, D.N.; Lefer, D.J. Modulation of leukocyte–endothelial interactions by reactive metabolites of oxygen and nitrogen: Relevance to ischemic heart disease. Free Radic. Biol. Med. 1998, 25, 404–433. [Google Scholar] [CrossRef]
- Dorweiler, B.; Pruefer, D.; Andrasi, T.B.; Maksan, S.M.; Schmiedt, W.; Neufang, A.; Vahl, C.F. Ischemia-Reperfusion Injury: Pathophysiology and Clinical Implications. Eur. J. Trauma Emerg. Surg. 2007, 33, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Lacy, F.; Gough, D.A.; Schmid-Schönbein, G.W. Role of xanthine oxidase in hydrogen peroxide production. Free Radic. Biol. Med. 1998, 25, 720–727. [Google Scholar] [CrossRef]
- Lum, H.; Barr, D.A.; Shaffer, J.R.; Gordon, R.J.; Ezrin, A.M.; Malik, A.B. Reoxygenation of endothelial cells increases permeability by oxidant-dependent mechanisms. Circ. Res. 1992, 70, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R.; Johnson, D.R.; Pober, J.S. Endothelial activation by hydrogen peroxide. Selective increases of intercellular adhesion molecule-1 and major histocompatibility complex class I. Am. J. Pathol. 1993, 142, 1598–1609. [Google Scholar] [PubMed]
- Lewis, M.S.; Whatley, R.E.; Cain, P.; McIntyre, T.M.; Prescott, S.M.; Zimmerman, G.A. Hydrogen peroxide stimulates the synthesis of platelet-activating factor by endothelium and induces endothelial cell-dependent neutrophil adhesion. J. Clin. Investig. 1988, 82, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Kvietys, P.R.; Granger, D.N. Role of reactive oxygen and nitrogen species in the vascular responses to inflammation. Free Radic. Biol. Med. 2012, 52, 556–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohman, A.W.; Billaud, M.; Isakson, B.E. Mechanisms of ATP release and signalling in the blood vessel wall. Cardiovasc. Res. 2012, 95, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, V.; Lu, B.; Rajakumar, S.; Cowan, P.J.; Dwyer, K.M. The CD39-adenosinergic axis in the pathogenesis of renal ischemia–reperfusion injury. Purinergic Signal. 2013, 9, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Cotrina, M.L.; Han, X.; Yu, H.; Bekar, L.; Blum, L.; Takano, T.; Tian, G.-F.; Goldman, S.A.; Nedergaard, M. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2009, 106, 12489–12493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G.; Knight, G.E. Cellular distribution and functions of P2 receptor subtypes in different systems. Int. Rev. Cytol. 2004, 240, 31–304. [Google Scholar] [PubMed]
- Harden, T.K.; Lazarowski, E.R.; Boucher, R.C. Release, metabolism and interconversion of adenine and uridine nucleotides: Implications for G protein-coupled P2 receptor agonist selectivity. Trends Pharmacol. Sci. 1997, 18, 43–46. [Google Scholar] [CrossRef]
- Weisman, G.A.; Erb, L.; Garrad, R.C.; Theiss, P.M.; Pérez, L.I.S.; Flores, R.V.; Berríos, C.S.; Méndez, Y.; González, F.A. P2Y nucleotide receptors in the immune system: Signaling by a P2Y2 receptor in U937 monocytes. Drug Dev. Res. 1998, 45, 222–228. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic signaling and vascular cell proliferation and death. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Cobbold, S.P.; Waldmann, H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2012, 171, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Köhler, D.; Eckle, T.; Kong, T.; Robson, S.C.; Colgan, S.P. Central role of Sp1-regulated CD39 in hypoxia/ischemia protection. Blood 2009, 113, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [PubMed]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Végran, F.; Hichami, A.; Ladoire, S.; Derangère, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Mishra, S.K.; Shukla, V.; Langer, D.; Gampe, K.; Grimm, I.; Delic, J.; Braun, N. Ecto-nucleotidases, molecular properties and functional impact. An. Real Acad. Nac. Farm. 2007, 73, 537–566. [Google Scholar]
- Linden, J. Molecular approach to adenosine receptors: Receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eltzschig, H.K.; Ibla, J.C.; Furuta, G.T.; Leonard, M.O.; Jacobson, K.A.; Enjyoji, K.; Robson, S.C.; Colgan, S.P. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: Role of ectonucleotidases and adenosine A2B receptors. J. Exp. Med. 2003, 198, 783–796. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Howie, D.; Nolan, K.F.; Agorogiannis, E.I.; Greaves, D.R.; Cobbold, S.P.; Waldmann, H. Generation of anti-inflammatory adenosine by leukocytes is regulated by TGF-β. Eur. J. Immunol. 2011, 41, 2955–2965. [Google Scholar] [CrossRef] [PubMed]
- Chisci, E.; De Giorgi, M.; Zanfrini, E.; Testasecca, A.; Brambilla, E.; Cinti, A.; Farina, L.; Kutryb Zajac, B.; Bugarin, C.; Villa, C.; et al. Simultaneous overexpression of human E5NT and ENTPD1 protects porcine endothelial cells against H2O2-induced oxidative stress and cytotoxicity in vitro. Free Radic. Biol. Med. 2017, 108, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Peyton, K.J.; Reyna, S.V.; Chapman, G.B.; Ensenat, D.; Liu, X.-M.; Wang, H.; Schafer, A.I.; Durante, W. Heme oxygenase-1-derived carbon monoxide is an autocrine inhibitor of vascular smooth muscle cell growth. Blood 2002, 99, 4443–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tobiasch, E.; Günther, L.; Bach, F.H. Heme oxygenase-1 protects pancreatic beta cells from apoptosis caused by various stimuli. J. Investig. Med. 2001, 49, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; Hanawa, H.; Ding, L.; Ota, Y.; Yoshida, K.; Toba, K.; Ogura, M.; Ito, H.; Kodama, M.; Aizawa, Y. Free heme is a danger signal inducing expression of proinflammatory proteins in cultured cells derived from normal rat hearts. Mol. Immunol. 2011, 48, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Bian, K.; Gao, Z.; Weisbrodt, N.; Murad, F. The nature of heme/iron-induced protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2003, 100, 5712–5717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennery, P.A. Signaling function of heme oxygenase proteins. Antioxid. Redox Signal. 2014, 20, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Nagababu, E.; Rifkind, J.M. Reaction of hydrogen peroxide with ferrylhemoglobin: Superoxide production and heme degradation. Biochemistry 2000, 39, 12503–12511. [Google Scholar] [CrossRef] [PubMed]
- Juckett, M.; Zheng, Y.; Yuan, H.; Pastor, T.; Antholine, W.; Weber, M.; Vercellotti, G. Heme and the endothelium. Effects of nitric oxide on catalytic iron and heme degradation by heme oxygenase. J. Biol. Chem. 1998, 273, 23388–23397. [Google Scholar] [CrossRef] [PubMed]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M.K. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 234–235, 249–263. [Google Scholar] [CrossRef]
- Kalish, H.R.; Latos-Grazyński, L.; Balch, A.L. Heme/Hydrogen peroxide reactivity: Formation of paramagnetic iron oxophlorin isomers treatment of iron porphyrins with hydrogen peroxide. J. Am. Chem. Soc. 2000, 122, 12478–12486. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. https://doi.org/10.3390/cells7100156
Di Marzo N, Chisci E, Giovannoni R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells. 2018; 7(10):156. https://doi.org/10.3390/cells7100156
Chicago/Turabian StyleDi Marzo, Noemi, Elisa Chisci, and Roberto Giovannoni. 2018. "The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells" Cells 7, no. 10: 156. https://doi.org/10.3390/cells7100156
APA StyleDi Marzo, N., Chisci, E., & Giovannoni, R. (2018). The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells, 7(10), 156. https://doi.org/10.3390/cells7100156