The Roles of Endo-Lysosomes in Unconventional Protein Secretion

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Lysosome or Endosome as a Secretory Compartment?
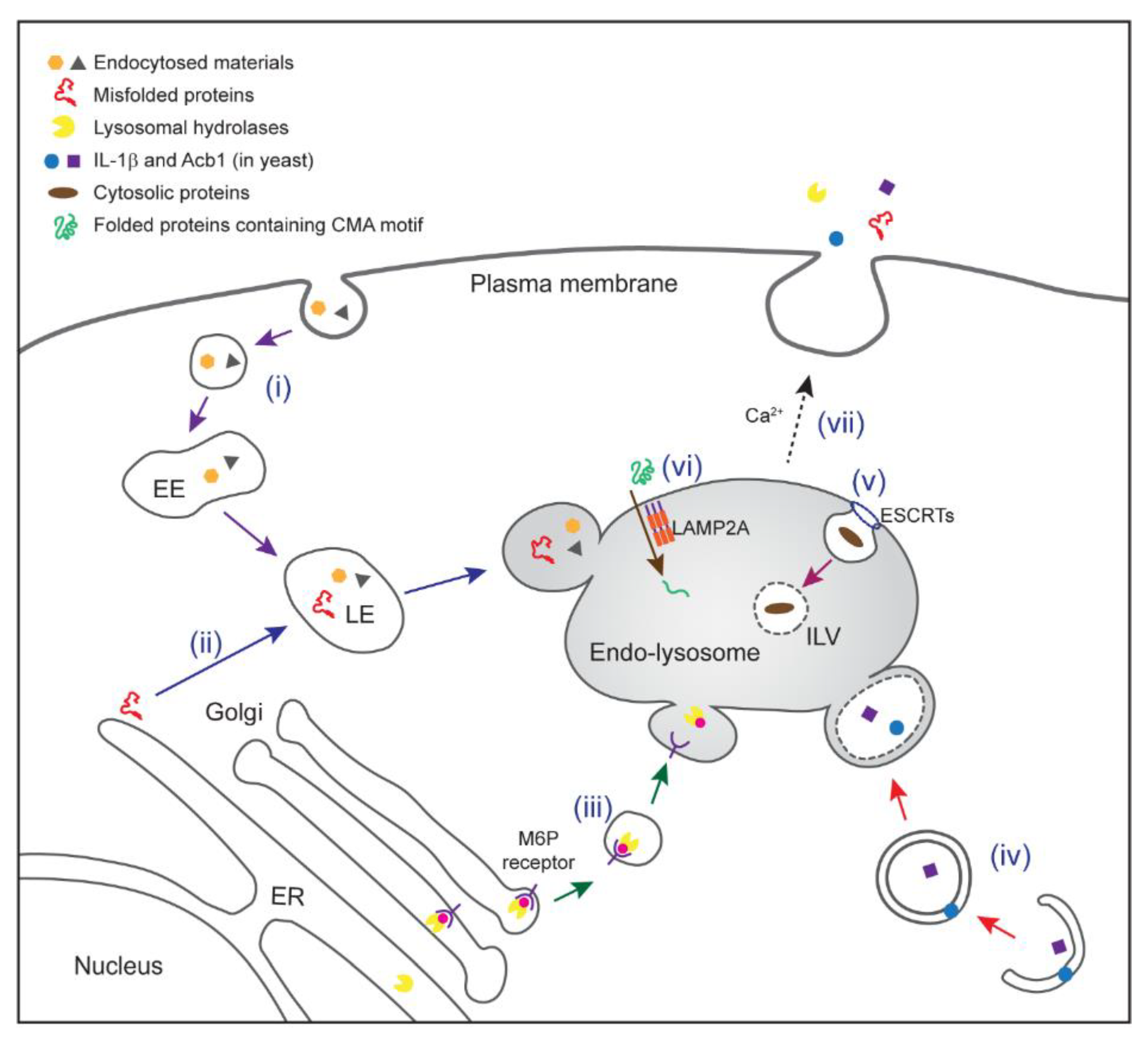
3. Protein Trafficking to Endo-Lysosomes
4. Protein Translocation across Endosome Membranes
5. Endo-Lysosome-Mediated Secretion and Human Diseases
6. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Rapoport, T.A.; Li, L.; Park, E. Structural and Mechanistic Insights into Protein Translocation. Annu. Rev. Cell Dev. Biol. 2017, 33, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.M.; Malkus, P.; Schekman, R. Out of the ER—Outfitters, escorts and guides. Trends Cell Biol. 1999, 9, 5–7. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Taguchi, M.; Kovacs, E.J.; Young, H.A.; Oppenheim, J.J. Intracellular localization of human monocyte associated interleukin 1 (IL 1) activity and release of biologically active IL 1 from monocytes by trypsin and plasmin. J. Immunol. 1986, 136, 2883–2891. [Google Scholar] [PubMed]
- Auron, P.E.; Warner, S.J.; Webb, A.C.; Cannon, J.G.; Bernheim, H.A.; McAdam, K.J.; Rosenwasser, L.J.; LoPreste, G.; Mucci, S.F.; Dinarello, C.A. Studies on the molecular nature of human interleukin 1. J. Immunol. 1987, 138, 1447–1456. [Google Scholar] [PubMed]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990, 9, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Wolff, S.M. The role of interleukin-1 in disease. N. Engl. J. Med. 1993, 328, 106–113. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, A.; Wilson, H.L.; Kiss-Toth, E.; Dower, S.K.; North, R.A.; Surprenant, A. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity 2001, 15, 825–835. [Google Scholar] [CrossRef]
- Qu, Y.; Franchi, L.; Nunez, G.; Dubyak, G.R. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J. Immunol. 2007, 179, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Andrei, C.; Dazzi, C.; Lotti, L.; Torrisi, M.R.; Chimini, G.; Rubartelli, A. The secretory route of the leaderless protein interleukin 1 beta involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 1999, 10, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Kenny, S.J.; Ge, L.; Xu, K.; Schekman, R. Translocation of interleukin-1 beta into a vesicle intermediate in autophagy-mediated secretion. Elife 2015, 4, e11205. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Martin-Sanchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.J.; Mortellaro, A.; et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Mason, I.J. The ins and outs of fibroblast growth factors. Cell 1994, 78, 547–552. [Google Scholar] [CrossRef]
- Akl, M.R.; Nagpal, P.; Ayoub, N.M.; Tai, B.; Prabhu, S.A.; Capac, C.M.; Gliksman, M.; Goy, A.; Suh, K.S. Molecular and clinical significance of fibroblast growth factor 2 (FGF2/bFGF) in malignancies of solid and hematological cancers for personalized therapies. Oncotarget 2016, 7, 44735–44762. [Google Scholar] [CrossRef] [PubMed]
- Schafer, T.; Zentgraf, H.; Zehe, C.; Brugger, B.; Bernhagen, J.; Nickel, W. Unconventional secretion of fibroblast growth factor 2 is mediated by direct translocation across the plasma membrane of mammalian cells. J. Biol. Chem. 2004, 279, 6244–6251. [Google Scholar] [CrossRef] [PubMed]
- Steringer, J.P.; Bleicken, S.; Andreas, H.; Zacherl, S.; Laussmann, M.; Temmerman, K.; Contreras, F.X.; Bharat, T.A.; Lechner, J.; Muller, H.M.; et al. Phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2)-dependent oligomerization of fibroblast growth factor 2 (FGF2) triggers the formation of a lipidic membrane pore implicated in unconventional secretion. J. Biol. Chem. 2012, 287, 27659–27669. [Google Scholar] [CrossRef] [PubMed]
- Steringer, J.P.; Lange, S.; Cujova, S.; Sachl, R.; Poojari, C.; Lolicato, F.; Beutel, O.; Muller, H.M.; Unger, S.; Coskun, U.; et al. Key steps in unconventional secretion of fibroblast growth factor 2 reconstituted with purified components. Elife 2017, 6, e28985. [Google Scholar] [CrossRef] [PubMed]
- Backhaus, R.; Zehe, C.; Wegehingel, S.; Kehlenbach, A.; Schwappach, B.; Nickel, W. Unconventional protein secretion: Membrane translocation of FGF-2 does not require protein unfolding. J. Cell Sci. 2004, 117, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- La Venuta, G.; Wegehingel, S.; Sehr, P.; Muller, H.M.; Dimou, E.; Steringer, J.P.; Grotwinkel, M.; Hentze, N.; Mayer, M.P.; Will, D.W.; et al. Small Molecule Inhibitors Targeting Tec Kinase Block Unconventional Secretion of Fibroblast Growth Factor 2. J. Biol. Chem. 2016, 291, 17787–17803. [Google Scholar] [CrossRef] [PubMed]
- Rayne, F.; Debaisieux, S.; Yezid, H.; Lin, Y.L.; Mettling, C.; Konate, K.; Chazal, N.; Arold, S.T.; Pugniere, M.; Sanchez, F.; et al. Phosphatidylinositol-(4,5)-bisphosphate enables efficient secretion of HIV-1 Tat by infected T-cells. EMBO J. 2010, 29, 1348–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitler, M.; Steringer, J.P.; Muller, H.M.; Mayer, M.P.; Nickel, W. HIV-Tat Protein Forms Phosphoinositide-dependent Membrane Pores Implicated in Unconventional Protein Secretion. J. Biol. Chem. 2015, 290, 21976–21984. [Google Scholar] [CrossRef] [PubMed]
- Prudovsky, I.; Bagala, C.; Tarantini, F.; Mandinova, A.; Soldi, R.; Bellum, S.; Maciag, T. The intracellular translocation of the components of the fibroblast growth factor 1 release complex precedes their assembly prior to export. J. Cell Biol. 2002, 158, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deora, A.B.; Kreitzer, G.; Jacovina, A.T.; Hajjar, K.A. An annexin 2 phosphorylation switch mediates p11-dependent translocation of annexin 2 to the cell surface. J. Biol. Chem. 2004, 279, 43411–43418. [Google Scholar] [CrossRef] [PubMed]
- Wegehingel, S.; Zehe, C.; Nickel, W. Rerouting of fibroblast growth factor 2 to the classical secretory pathway results in post-translational modifications that block binding to heparan sulfate proteoglycans. FEBS Lett. 2008, 582, 2387–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecceu, F.; Dousset, P.; Shire, D.; Cavrois, E.; Marchese, E.; Ferrara, P.; Kaghad, M.; Dumont, X.; Lupker, J. Human interleukin 1 beta fused to the human growth hormone signal peptide is N-glycosylated and secreted by Chinese hamster ovary cells. Gene 1991, 97, 253–258. [Google Scholar] [CrossRef]
- Fleer, R.; Chen, X.J.; Amellal, N.; Yeh, P.; Fournier, A.; Guinet, F.; Gault, N.; Faucher, D.; Folliard, F.; Fukuhara, H.; et al. High-level secretion of correctly processed recombinant human interleukin-1 beta in Kluyveromyces lactis. Gene 1991, 107, 285–295. [Google Scholar] [CrossRef]
- Kinseth, M.A.; Anjard, C.; Fuller, D.; Guizzunti, G.; Loomis, W.F.; Malhotra, V. The Golgi-associated protein GRASP is required for unconventional protein secretion during development. Cell 2007, 130, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Cabral, M.; Anjard, C.; Malhotra, V.; Loomis, W.F.; Kuspa, A. Unconventional secretion of AcbA in Dictyostelium discoideum through a vesicular intermediate. Eukaryotic Cell 2010, 9, 1009–1017. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curwin, A.J.; Brouwers, N.; Alonso, Y.A.M.; Teis, D.; Turacchio, G.; Parashuraman, S.; Ronchi, P.; Malhotra, V. ESCRT-III drives the final stages of CUPS maturation for unconventional protein secretion. Elife 2016, 5, e16299. [Google Scholar] [CrossRef] [PubMed]
- Bruns, C.; McCaffery, J.M.; Curwin, A.J.; Duran, J.M.; Malhotra, V. Biogenesis of a novel compartment for autophagosome-mediated unconventional protein secretion. J. Cell Biol. 2011, 195, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of DeltaF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.H.; Gee, H.Y.; Kim, Y.; Piao, H.; Kim, J.; Kang, C.M.; Lee, G.; Mook-Jung, I.; Lee, Y.; Cho, J.W.; et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy 2018, 14, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Burdett, I.; Hughes, R.C. Secretion of the baby hamster kidney 30-kDa galactose-binding lectin from polarized and nonpolarized cells: A pathway independent of the endoplasmic reticulum-Golgi complex. Exp. Cell Res. 1993, 207, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Lukyanov, P.; Furtak, V.; Ochieng, J. Galectin-3 interacts with membrane lipids and penetrates the lipid bilayer. Biochem. Biophys. Res. Commun. 2005, 338, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.C.; Honjo, Y.; Nangia-Makker, P.; Hogan, V.; Mazurak, N.; Bresalier, R.S.; Raz, A. The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res. 1999, 59, 6239–6245. [Google Scholar] [PubMed]
- Schneider, D.; Greb, C.; Koch, A.; Straube, T.; Elli, A.; Delacour, D.; Jacob, R. Trafficking of galectin-3 through endosomal organelles of polarized and non-polarized cells. Eur. J. Cell Biol. 2010, 89, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.N.; Zheng, D.; Sabbagh, J.J.; Martin, M.D.; Chaput, D.; Darling, A.; Trotter, J.H.; Stothert, A.R.; Nordhues, B.A.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
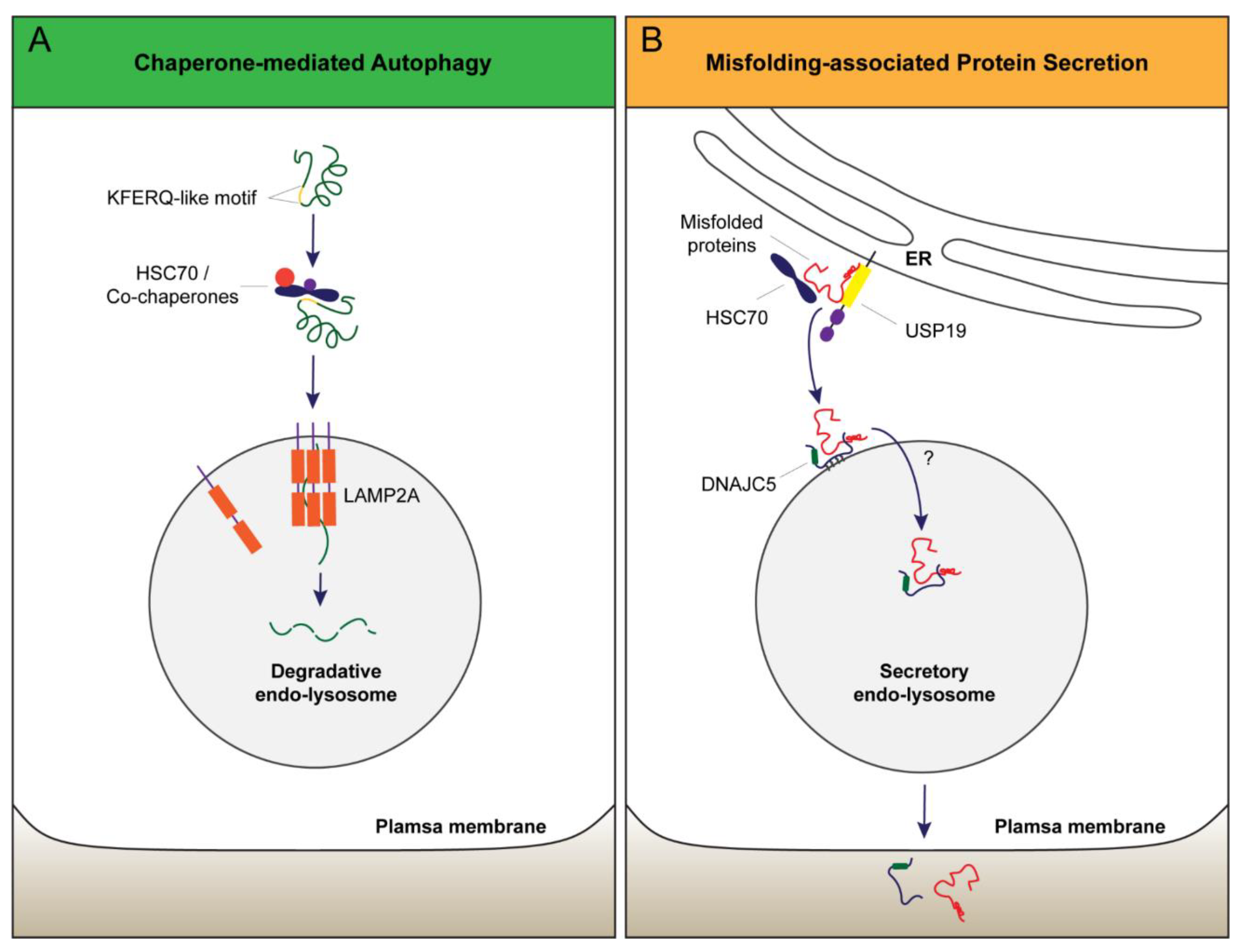
- Xu, Y.; Cui, L.; Dibello, A.; Wang, L.; Lee, J.; Saidi, L.; Lee, J.G.; Ye, Y. DNAJC5 facilitates USP19-dependent unconventional secretion of misfolded cytosolic proteins. Cell Discov. 2018, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y. Regulation of protein homeostasis by unconventional protein secretion in mammalian cells. Semin. Cell Dev. Biol. 2018, 83, 29–35. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillay, C.S.; Elliott, E.; Dennison, C. Endolysosomal proteolysis and its regulation. Biochem. J. 2002, 363, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, J.D.; Palade, G.E. Intracellular transport of secretory proteins in the pancreatic exocrine cell. II. Transport to condensing vacuoles and zymogen granules. J. Cell Biol. 1967, 34, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Vaes, G. On the mechanisms of bone resorption. The action of parathyroid hormone on the excretion and synthesis of lysosomal enzymes and on the extracellular release of acid by bone cells. J. Cell Biol. 1968, 39, 676–697. [Google Scholar] [CrossRef] [PubMed]
- Doty, S.B.; Schofield, B.H. Electron microscopic localization of hydrolytic enzymes in osteoclasts. Histochem. J. 1972, 4, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Gothlin, G.; Ericsson, J.L. Fine structural localization of acid phosphomonoesterase in the brush border region of osteoclasts. Histochimie 1971, 28, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Davison, I.; Shankar, G.; Horton, M.A.; Mason, W.T. Integrin-dependent mobilization of intracellular calcium ions in osteoclasts. A possible role in the regulation of the secretion of protons and lysosomal enzymes. Ann. N. Y. Acad. Sci. 1994, 710, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Muller, M. Secretion of acid hydrolases and its intracellular source in Tetrahymena pyriformis. J. Cell Biol. 1972, 52, 478–487. [Google Scholar] [CrossRef] [PubMed]
- Peters, P.J.; Borst, J.; Oorschot, V.; Fukuda, M.; Krahenbuhl, O.; Tschopp, J.; Slot, J.W.; Geuze, H.J. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med. 1991, 173, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjeldsen, L.; Bainton, D.F.; Sengelov, H.; Borregaard, N. Structural and functional heterogeneity among peroxidase-negative granules in human neutrophils: Identification of a distinct gelatinase-containing granule subset by combined immunocytochemistry and subcellular fractionation. Blood 1993, 82, 3183–3191. [Google Scholar] [PubMed]
- Burkhardt, J.K.; Hester, S.; Lapham, C.K.; Argon, Y. The lytic granules of natural killer cells are dual-function organelles combining secretory and pre-lysosomal compartments. J. Cell Biol. 1990, 111, 2327–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blott, E.J.; Griffiths, G.M. Secretory lysosomes. Nat. Rev. Mol. Cell Biol. 2002, 3, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Stutchfield, J.; Cockcroft, S. Correlation between secretion and phospholipase D activation in differentiated HL60 cells. Biochem. J. 1993, 293 Pt 3, 649–655. [Google Scholar] [CrossRef]
- Fensome, A.; Cunningham, E.; Prosser, S.; Tan, S.K.; Swigart, P.; Thomas, G.; Hsuan, J.; Cockcroft, S. ARF and PITP restore GTP gamma S-stimulated protein secretion from cytosol-depleted HL60 cells by promoting PIP2 synthesis. Curr. Biol. 1996, 6, 730–738. [Google Scholar] [CrossRef]
- Jones, D.H.; Bax, B.; Fensome, A.; Cockcroft, S. ADP ribosylation factor 1 mutants identify a phospholipase D effector region and reveal that phospholipase D participates in lysosomal secretion but is not sufficient for recruitment of coatomer I. Biochem. J. 1999, 341 Pt 1, 185–192. [Google Scholar] [CrossRef]
- Rodriguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, J.K.; Andrews, N.W.; Simon, S.M. Membrane proximal lysosomes are the major vesicles responsible for calcium-dependent exocytosis in nonsecretory cells. J. Cell Biol. 2002, 159, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.C.; Vacca, F.; Gruenberg, J. Endosome maturation, transport and functions. Semin. Cell Dev. Biol. 2014, 31, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 2018, 217, 3127–3139. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.T.; Xie, Y.X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.H. Revisiting LAMP1 as a marker for degradative autophagy-lysosomal organelles in the nervous system. Autophagy 2018, 14, 1472–1474. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, J.; Bassaganyas, L.; Lepreux, S.; Chiritoiu, M.; Costet, P.; Ripoche, J.; Malhotra, V.; Schekman, R. Unconventional secretion of FABP4 by endosomes and secretory lysosomes. J. Cell Biol. 2018, 217, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.; Hoflack, B.; Simons, K.; Mellman, I.; Kornfeld, S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 1988, 52, 329–341. [Google Scholar] [CrossRef]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Reczek, D.; Schwake, M.; Schroder, J.; Hughes, H.; Blanz, J.; Jin, X.; Brondyk, W.; Van Patten, S.; Edmunds, T.; Saftig, P. LIMP-2 is a receptor for lysosomal mannose-6-phosphate-independent targeting of beta-glucocerebrosidase. Cell 2007, 131, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ren, J.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Lysosome sorting of beta-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat. Commun. 2014, 5, 4321. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Hannan, L.A.; Marks, M.S.; Stevens, T.H.; Schroer, T.A. Fundamental mechanisms deliver the Nobel Prize to Ohsumi. Traffic 2017, 18, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Archiv. B Cell Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Ehrenreich, B.A.; Cohn, Z.A. The fate of peptides pinocytosed by macrophages in vitro. J. Exp. Med. 1969, 129, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Auteri, J.S.; Okada, A.; Bochaki, V.; Dice, J.F. Regulation of intracellular protein degradation in IMR-90 human diploid fibroblasts. J. Cell. Physiol. 1983, 115, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995, 269, C1200–C1208. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S.; Chiang, H.L.; Goldberg, A.L.; Dice, J.F. Proteins containing peptide sequences related to Lys-Phe-Glu-Arg-Gln are selectively depleted in liver and heart, but not skeletal muscle, of fasted rats. Biochem. J. 1991, 275 Pt 1, 165–169. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Chaperone-mediated autophagy: Dice’s ‘wild’ idea about lysosomal selectivity. Nat. Rev. Mol. Cell Biol. 2011, 12, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. A receptor for the selective uptake and degradation of proteins by lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Dice, J.F. Peptide sequences that target proteins for enhanced degradation during serum withdrawal. J. Biol. Chem. 1988, 263, 6797–6805. [Google Scholar] [PubMed]
- Lee, J.; Xu, Y.; Zhang, T.; Cui, L.; Saidi, L.; Ye, Y. Secretion of misfolded cytosolic proteins from mammalian cells is independent of chaperone-mediated autophagy. J. Biol. Chem. 2018, 293, 14359–14370. [Google Scholar] [CrossRef] [PubMed]
- Noskova, L.; Stranecky, V.; Hartmannova, H.; Pristoupilova, A.; Baresova, V.; Ivanek, R.; Hulkova, H.; Jahnova, H.; van der Zee, J.; Staropoli, J.F.; et al. Mutations in DNAJC5, encoding cysteine-string protein alpha, cause autosomal-dominant adult-onset neuronal ceroid lipofuscinosis. Am. J. Hum. Genet. 2011, 89, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Benitez, B.A.; Alvarado, D.; Cai, Y.; Mayo, K.; Chakraverty, S.; Norton, J.; Morris, J.C.; Sands, M.S.; Goate, A.; Cruchaga, C. Exome-sequencing confirms DNAJC5 mutations as cause of adult neuronal ceroid-lipofuscinosis. PLoS ONE 2011, 6, e26741. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Loganathan, K.; Sekiguchi, M.; Matsuba, Y.; Hui, K.; Tsubuki, S.; Tanaka, M.; Iwata, N.; Saito, T.; Saido, T.C. Abeta secretion and plaque formation depend on autophagy. Cell Rep. 2013, 5, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.; et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 2012, 287, 3842–3849. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Garcia-Garcia, E.; Gomez-Ramos, A.; Falcon-Perez, J.M.; Diaz-Hernandez, M.; Hernandez, F.; Avila, J. Tau overexpression results in its secretion via membrane vesicles. Neuro-Degener. Dis. 2012, 10, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kugler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dujardin, S.; Begard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.C.; Bousset, L.; Melki, R.; et al. Ectosomes: A new mechanism for non-exosomal secretion of tau protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Mohamed, N.V.; Desjardins, A.; Lippe, R.; Fon, E.A.; Leclerc, N. Rab7A regulates tau secretion. J. Neurochem. 2017, 141, 592–605. [Google Scholar] [CrossRef] [PubMed]
- Katsinelos, T.; Zeitler, M.; Dimou, E.; Karakatsani, A.; Muller, H.M.; Nachman, E.; Steringer, J.P.; Ruiz de Almodovar, C.; Nickel, W.; Jahn, T.R. Unconventional Secretion Mediates the Trans-cellular Spreading of Tau. Cell Rep. 2018, 23, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Jeong, H.; Krainc, D. Mutant Huntingtin Is Secreted via a Late Endosomal/Lysosomal Unconventional Secretory Pathway. J. Neurosci. 2017, 37, 9000–9012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapper, H.; Sundler, R. Role of lysosomal and cytosolic pH in the regulation of macrophage lysosomal enzyme secretion. Biochem. J. 1990, 272, 407–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.; Ye, Y. The Roles of Endo-Lysosomes in Unconventional Protein Secretion. Cells 2018, 7, 198. https://doi.org/10.3390/cells7110198
Lee J, Ye Y. The Roles of Endo-Lysosomes in Unconventional Protein Secretion. Cells. 2018; 7(11):198. https://doi.org/10.3390/cells7110198
Chicago/Turabian StyleLee, Juhyung, and Yihong Ye. 2018. "The Roles of Endo-Lysosomes in Unconventional Protein Secretion" Cells 7, no. 11: 198. https://doi.org/10.3390/cells7110198
APA StyleLee, J., & Ye, Y. (2018). The Roles of Endo-Lysosomes in Unconventional Protein Secretion. Cells, 7(11), 198. https://doi.org/10.3390/cells7110198