The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. ChIP-seq
2.3. RNA Purification and RNA-seq
2.4. Peak Calling and Peak Analysis
2.5. Calculation of ChIP-seq Coverage and Signal Distribution Across Specific Genomic Location
2.6. Correlation Analysis and Combinatorial Pattern of Heterochromatin Markers
2.7. RNA-seq Analysis
2.8. Distribution of Heterochromatin Signals Across High and Low Abundant Genes
3. Results
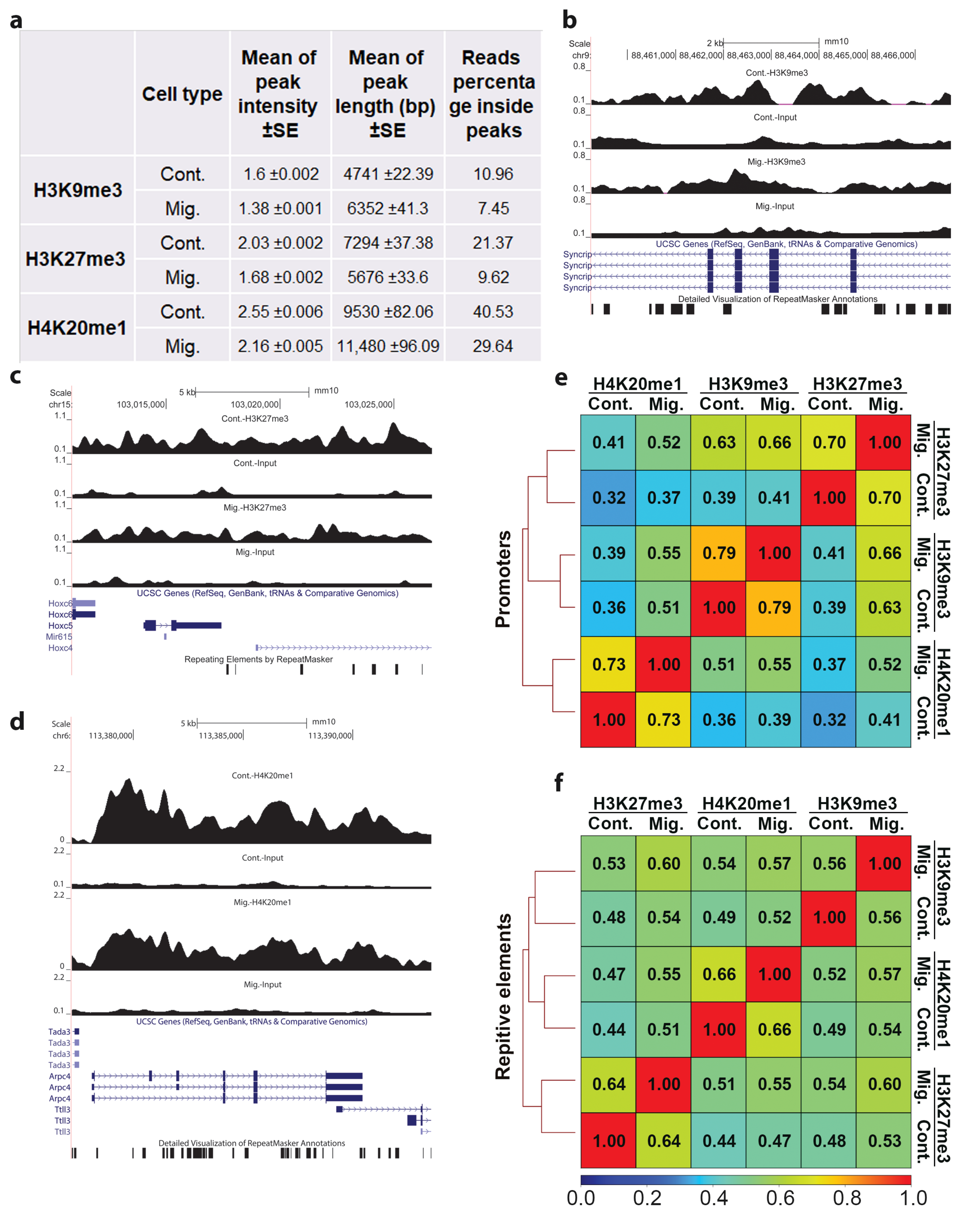
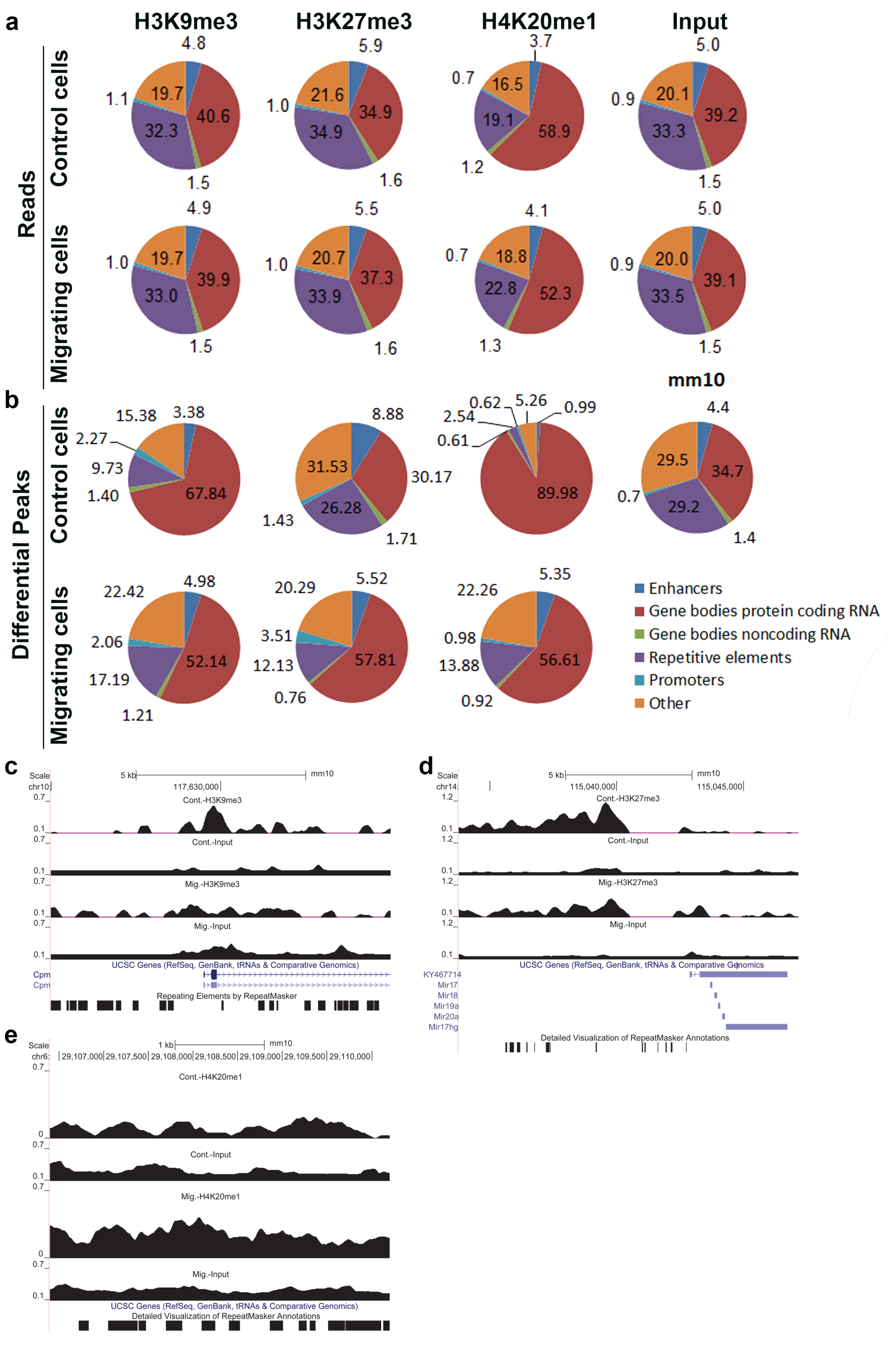
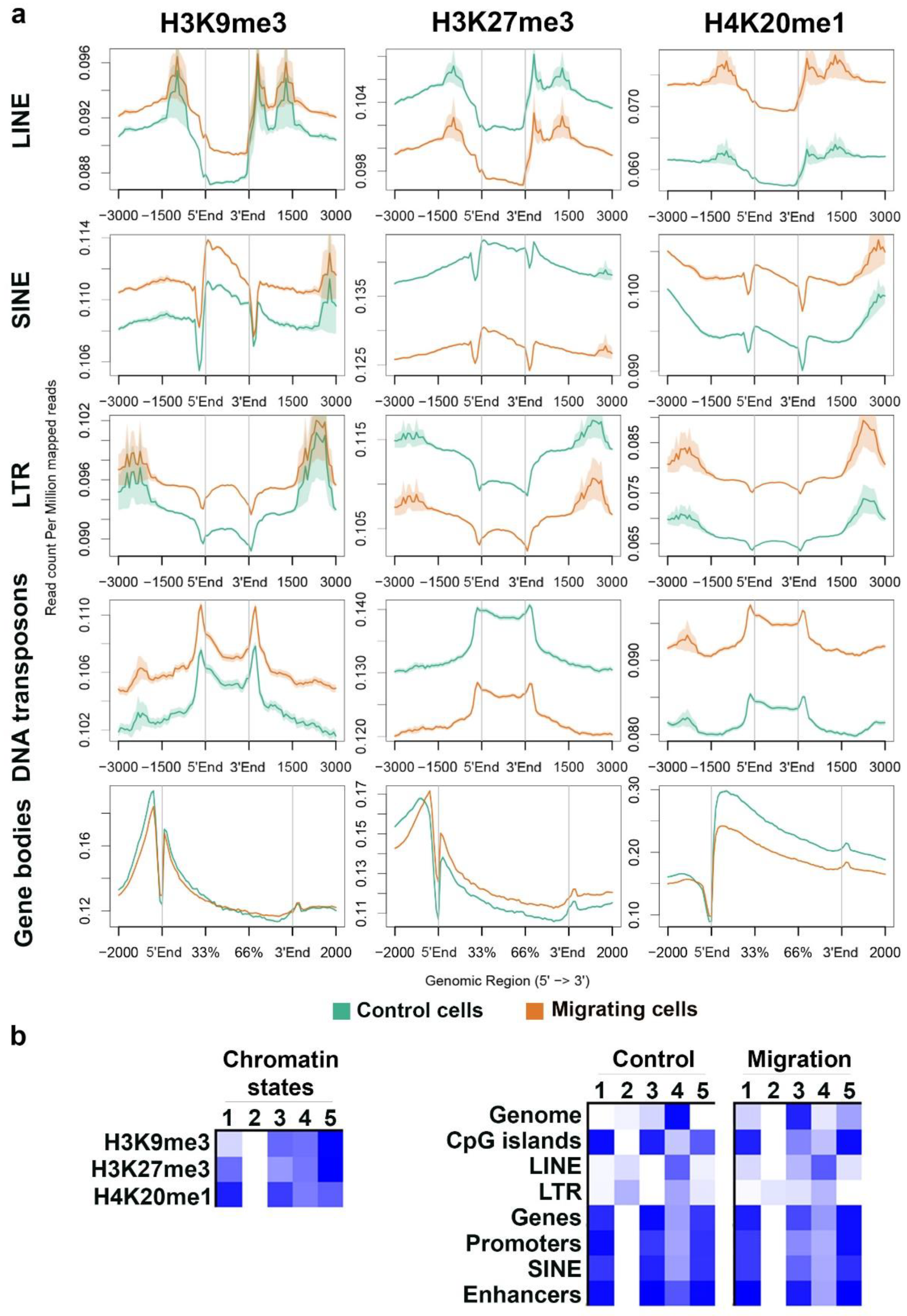
3.1. Migration-Induced Changes in the Genomic Distribution of H3K9me3, H3K27me3, and H4K20me1
3.2. Migration-Induced Transcriptome Changes
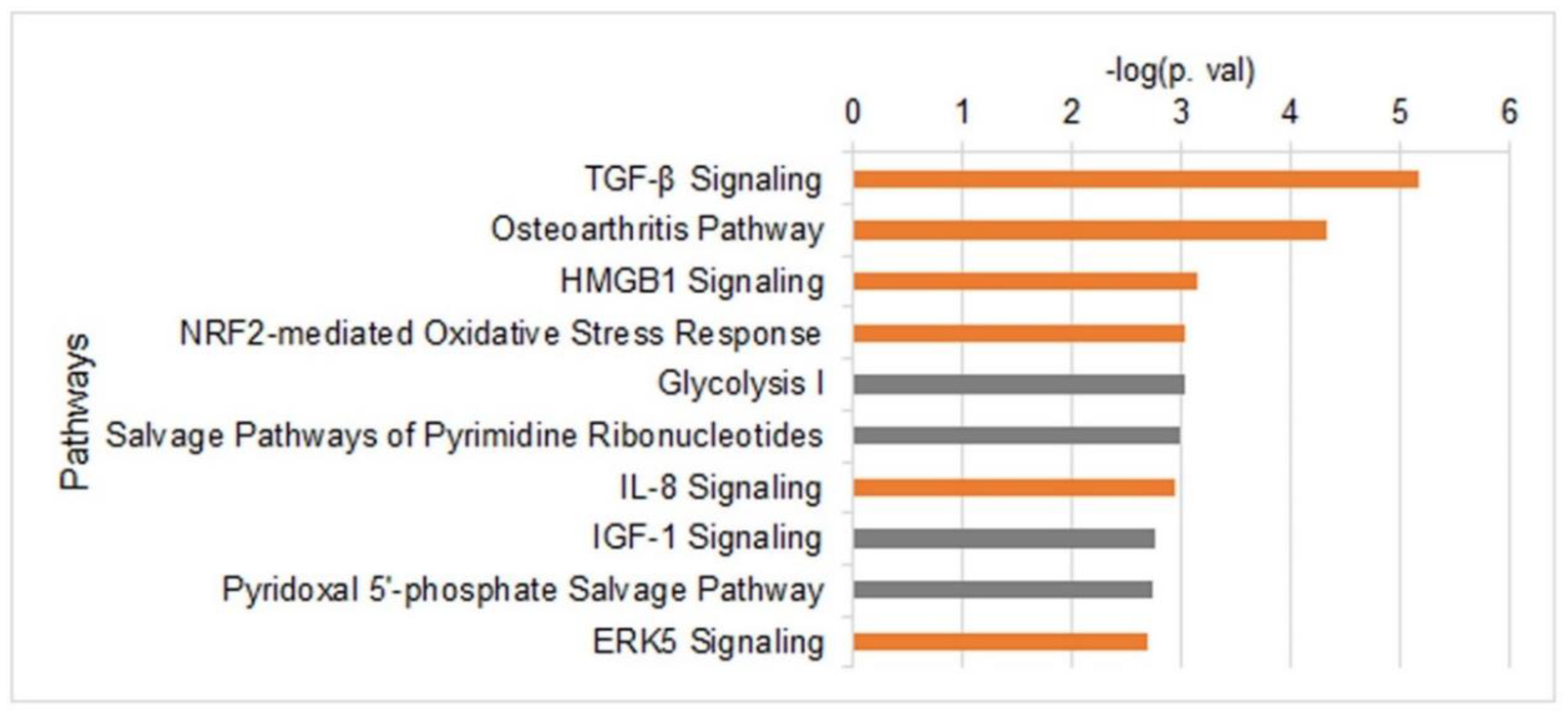
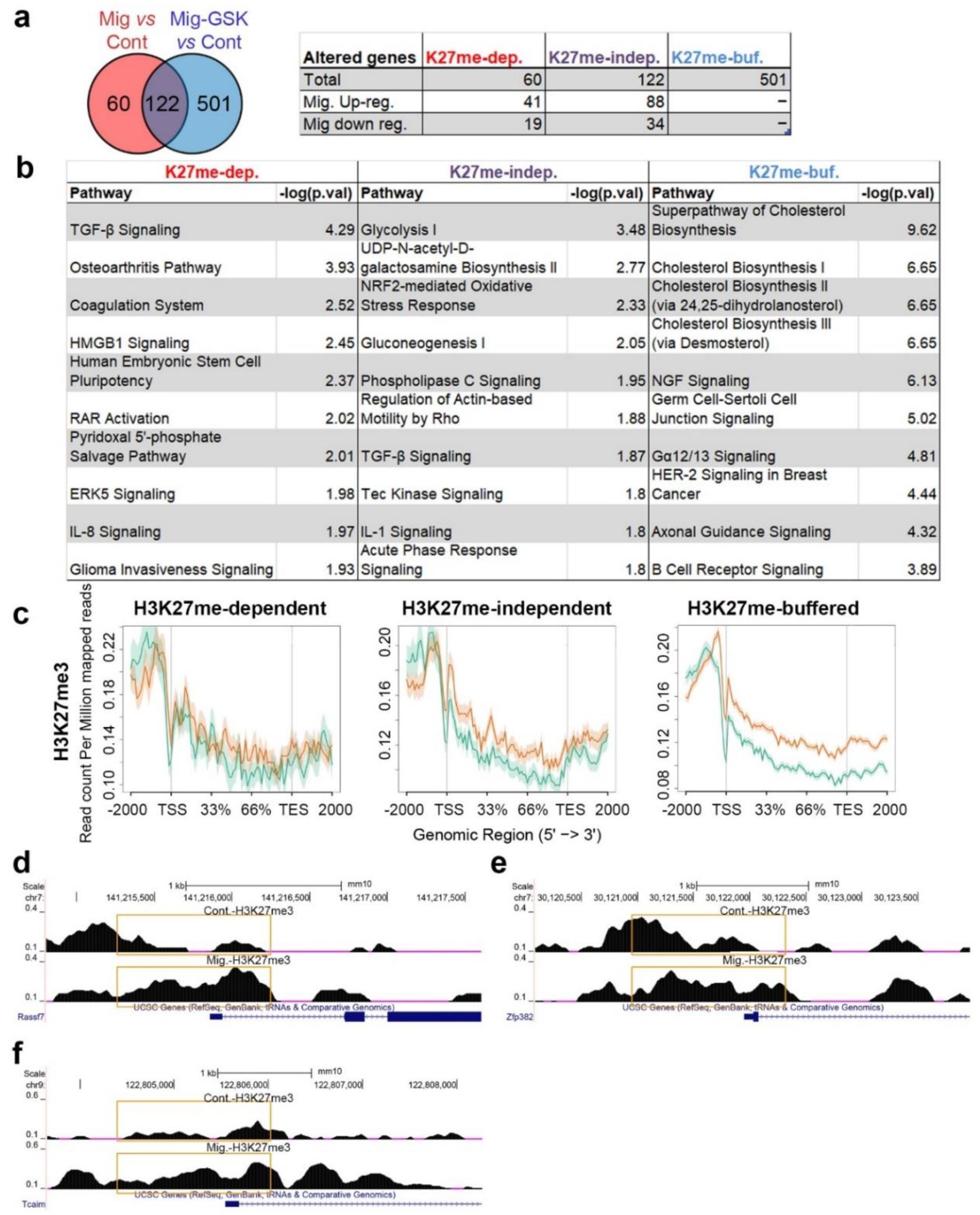
3.3. H3K27me-Dependent Transcriptome Changes upon Induction of Migration
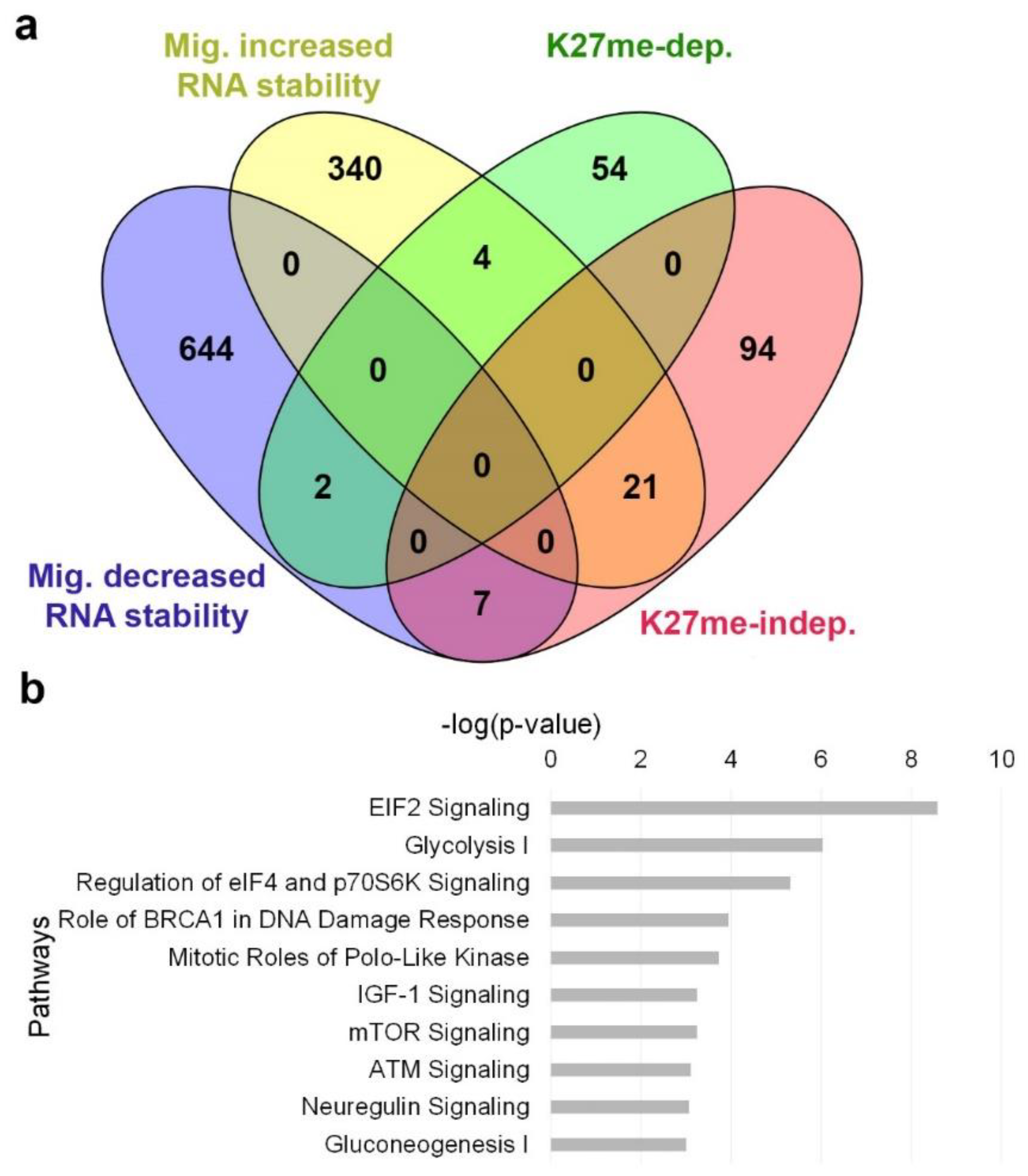
3.4. H3K27me-Dependent Genes Are Less Prone to RNA Stability Control
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Data Availability
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S. Microtubules in Cell Migration. Annu. Rev. Cell Dev. Biol. 2013, 29, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Rottner, K.; Stradal, T.E. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Ouderkirk, J.L.; Krendel, M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis: Non-Muscle Myosins and Cancer. Cytoskeleton 2014, 71, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Gerlitz, G.; Bustin, M. The role of chromatin structure in cell migration. Trends Cell Biol. 2011, 21, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinty, R.K.; Tan, S. Histone, Nucleosome, and Chromatin Structure. In Fundamentals of Chromatin; Workman, J.L., Abmayr, S.M., Eds.; Springer: New York, NY, USA, 2014; pp. 1–28. [Google Scholar]
- Mozzetta, C.; Boyarchuk, E.; Pontis, J.; Ait-Si-Ali, S. Sound of silence: The properties and functions of repressive Lys methyltransferases. Nat. Rev. Mol. Cell Biol. 2015, 16, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schofer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rougeulle, C.; Chaumeil, J.; Sarma, K.; Allis, C.D.; Reinberg, D.; Avner, P.; Heard, E. Differential histone H3 Lys-9 and Lys-27 methylation profiles on the X. chromosome. Mol. Cell Biol. 2004, 24, 5475–5484. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Oda, H.; Shen, S.S.; Reinberg, D. PR-Set7 and H4K20me1: At the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012, 26, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kohlmaier, A.; Savarese, F.; Lachner, M.; Martens, J.; Jenuwein, T.; Wutz, A. A Chromosomal Memory Triggered by Xist Regulates Histone Methylation in X. Inactivation. PLoS Biol. 2004, 2, e171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlitz, G.; Bustin, M. Efficient cell migration requires global chromatin condensation. J. Cell Sci. 2010, 123, 2207–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlitz, G.; Livnat, I.; Ziv, C.; Yarden, O.; Bustin, M.; Reiner, O. Migration cues induce chromatin alterations. Traffic 2007, 8, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Maizels, Y.; Elbaz, A.; Hernandez-Vicens, R.; Sandrusy, O.; Rosenberg, A.; Gerlitz, G. Increased chromatin plasticity supports enhanced metastatic potential of mouse melanoma cells. Exp. Cell Res. 2017, 357, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Cook, P.C.; Zindy, E.; Williams, C.J.; Jowitt, T.A.; Streuli, C.H.; MacDonald, A.S.; Redondo-Muñoz, J. Integrin α4β1 controls G9a activity that regulates epigenetic changes and nuclear properties required for lymphocyte migration. Nucleic Acids Res. 2016, 44, 3031–3044. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Luo, Q.; Chen, Z.; Shi, Y.; Ju, Y.; Yang, L.; Song, G. Increased nuclear stiffness via FAK-ERK1/2 signaling is necessary for synthetic mechano-growth factor E peptide-induced tenocyte migration. Sci. Rep. 2016, 6, 18809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Luo, Q.; Sun, J.; Ju, Y.; Morita, Y.; Song, G. Chromatin organization regulated by EZH2-mediated H3K27me3 is required for OPN-induced migration of bone marrow-derived mesenchymal stem cells. Int. J. Biochem. Cell Biol. 2018, 96, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; et al. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar] [CrossRef] [PubMed]
- Kottakis, F.; Polytarchou, C.; Foltopoulou, P.; Sanidas, I.; Kampranis, S.C.; Tsichlis, P.N. FGF-2 Regulates Cell Proliferation, Migration, and Angiogenesis through an NDY1/KDM2B-miR-101-EZH2 Pathway. Mol. Cell 2011, 43, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chin, L.K.; Bourouina, T.; Liu, A.Q.; VanDongen, A.M.J. Nuclear deformation during breast cancer cell transmigration. Lab Chip 2012, 12, 3774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokura, K.; Sun, L.; Bedford, M.T.; Fang, J. Methyl-H3K9-binding protein MPP8 mediates E-cadherin gene silencing and promotes tumour cell motility and invasion. EMBO J. 2010, 29, 3673–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Wang, C.; Yang, J.; Guo, Y.; Wu, Y.; Li, X. Metformin inhibits SUV39H1-mediated migration of prostate cancer cells. Oncogenesis 2017, 6, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spyropoulou, A.; Gargalionis, A.; Dalagiorgou, G.; Adamopoulos, C.; Papavassiliou, K.A.; Lea, R.W.; Piperi, C.; Papavassiliou, A.G. Role of Histone Lysine Methyltransferases SUV39H1 and SETDB1 in Gliomagenesis: Modulation of Cell Proliferation, Migration, and Colony Formation. Neuromol. Med. 2014, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Girard, N.; Bazille, C.; Lhuissier, E.; Benateau, H.; Llombart-Bosch, A.; Boumediene, K.; Bauge, C. 3-Deazaneplanocin A. (DZNep), an Inhibitor of the Histone Methyltransferase EZH2, Induces Apoptosis and Reduces Cell Migration in Chondrosarcoma Cells. PLoS ONE 2014, 9, e98176. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Balasubramanian, S.; Kerr, C.; Huang, J.M.; Eckert, R.L. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015, 36, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio, O.; Geisberg, J.V.; Sekinger, E.; Yang, A.; Moqtaderi, Z.; Struhl, K. Chromatin Immunoprecipitation for Determining the Association of Proteins with Specific Genomic Sequences In Vivo. In Current Protocols in Molecular Biology; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Andrews, S. FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 November 2010).
- Krueger, F. Trim_galore. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 19 October 2012).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 24 November 2009).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard. Available online: https://broadinstitute.github.io/picard/ (accessed on 9 November 2018).
- Xu, S.; Grullon, S.; Ge, K.; Peng, W. Spatial Clustering for Identification of ChIP-Enriched Regions (SICER) to Map Regions of Histone Methylation Patterns in Embryonic Stem Cells. Methods Mol. Biol. 2014, 1150, 97–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinform. Oxf. Engl. 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Tyner, C.; Barber, G.P.; Casper, J.; Clawson, H.; Diekhans, M.; Eisenhart, C.; Fischer, C.M.; Gibson, D.; Gonzalez, J.N.; Guruvadoo, L.; et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res. 2017, 45, D626–D634. [Google Scholar] [CrossRef] [PubMed]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B. cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Shao, N.; Liu, X.; Nestler, E. ngs. plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genom. 2014, 15, 284. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Ryan, D.P.; Grüning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dündar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.; Kellis, M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinform. Oxf. Engl. 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Reimers, M.; Carey, V.J. Bioconductor: An Open Source Framework for Bioinformatics and Computational Biology. In Methods in Enzymology; DNA Microarrays, Part B: Databases and Statistics; Academic Press: Cambridge, MA, USA, 2006; pp. 119–134. [Google Scholar]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Gu, X.; Yi, S. Ingenuity Pathway Analysis of Gene Expression Profiles in Distal Nerve Stump following Nerve Injury: Insights into Wallerian Degeneration. Front. Cell. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.-C.; Park, A.Y.; Guan, J.-L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Gabellini, C.; Trisciuoglio, D.; Desideri, M.; Candiloro, A.; Ragazzoni, Y.; Orlandi, A.; Zupi, G.; Del Bufalo, D. Functional activity of CXCL8 receptors, CXCR1 and CXCR2, on human malignant melanoma progression. Eur. J. Cancer 2009, 45, 2618–2627. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, Y.; Manegold, P.C.; Hong, Y.K.; Zhang, W.; Pohl, A.; Lurje, G.; Winder, T.; Yang, D.; LaBonte, M.J.; Wilson, P.M.; et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 2011, 128, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Pang, M.-F.; Georgoudaki, A.-M.; Lambut, L.; Johansson, J.; Tabor, V.; Hagikura, K.; Jin, Y.; Jansson, M.; Alexander, J.S.; Nelson, C.M.; et al. TGF-β1-induced EMT promotes targeted migration of breast cancer cells through the lymphatic system by the activation of CCR7/CCL21-mediated chemotaxis. Oncogene 2016, 35, 748–760. [Google Scholar] [CrossRef] [PubMed]
- Simões, A.E.S.; Rodrigues, C.M.P.; Borralho, P.M. The MEK5/ERK5 signalling pathway in cancer: A promising novel therapeutic target. Drug Discov. Today 2016, 21, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Bird, R.P. The Emerging Role of Vitamin B6 in Inflammation and Carcinogenesis. In Advances in Food and Nutrition Research; Elsevier: Amsterdam, The Netherlands, 2018; pp. 151–194. [Google Scholar]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: An interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Dayem, M.A.; Moreilhon, C.; Turchi, L.; Magnone, V.; Christen, R.; Ponzio, G.; Barbry, P. Early Gene Expression in Wounded Human Keratinocytes Revealed by DNA Microarray Analysis. Comp. Funct. Genom. 2003, 4, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Demuth, T.; Rennert, J.L.; Hoelzinger, D.B.; Reavie, L.B.; Nakada, M.; Beaudry, C.; Nakada, S.; Anderson, E.M.; Henrichs, A.N.; McDonough, W.S.; et al. Glioma cells on the run—The migratory transcriptome of 10 human glioma cell lines. BMC Genom. 2008, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Fitsialos, G.; Chassot, A.-A.; Turchi, L.; Dayem, M.A.; LeBrigand, K.; Moreilhon, C.; Meneguzzi, G.; Buscà, R.; Mari, B.; Barbry, P.; et al. Transcriptional Signature of Epidermal Keratinocytes Subjected to in vitro Scratch Wounding Reveals Selective Roles for ERK1/2, p38, and Phosphatidylinositol 3-Kinase Signaling Pathways. J. Biol. Chem. 2007, 282, 15090–15102. [Google Scholar] [CrossRef] [PubMed]
- Vakoc, C.R.; Mandat, S.A.; Olenchock, B.A.; Blobel, G.A. Histone H3 lysine 9 methylation and HP1gamma are associated with transcription elongation through mammalian chromatin. Mol. Cell 2005, 19, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, A.B.; Roelofsen, T.; Pennings, S.W.C.; Martens, J.H.A.; Jenuwein, T.; Stunnenberg, H.G. Histone modification patterns associated with the human X. chromosome. EMBO Rep. 2006. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Ebihara, A.; Kajiho, H.; Kontani, K.; Nishina, H.; Katada, T. RASSF7 negatively regulates pro-apoptotic JNK signaling by inhibiting the activity of phosphorylated-MKK7. Cell Death Differ. 2011, 18, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Nätt, D.; Johansson, I.; Faresjö, T.; Ludvigsson, J.; Thorsell, A. High cortisol in 5-year-old children causes loss of DNA methylation in SINE retrotransposons: A possible role for ZNF263 in stress-related diseases. Clin. Epigenetics 2015, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Xiang, T.; Li, S.; Ye, L.; Feng, Y.; Pei, L.; Li, L.; Wang, X.; Sun, R.; Ren, G.; et al. The novel 19q13 KRAB zinc-finger tumour suppressor ZNF382 is frequently methylated in oesophageal squamous cell carcinoma and antagonises Wnt/β-catenin signalling. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.Z.; Schlickeiser, S.; Jürchott, K.; Akyuez, L.; Schumann, J.; Appelt, C.; Vogt, K.; Schröder, M.; Vaeth, M.; Berberich-Siebelt, F.; et al. TCAIM decreases T cell priming capacity of dendritic cells by inhibiting TLR-induced Ca2+ influx and IL-2 production. J. Immunol. 2015, 201, 1400713. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, T.; Rochman, M.; Taher, L.; Dimitriadis, E.K.; Nagashima, K.; Anderson, S.; Bustin, M. Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat. Commun. 2015, 6, 6138. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D.; Liu, P.Z.; Banigan, E.J.; Almassalha, L.M.; Backman, V.; Adam, S.A.; Goldman, R.D.; Marko, J.F. Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol. Biol. Cell 2018, 29, 220–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segal, T.; Salmon-Divon, M.; Gerlitz, G. The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations. Cells 2018, 7, 205. https://doi.org/10.3390/cells7110205
Segal T, Salmon-Divon M, Gerlitz G. The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations. Cells. 2018; 7(11):205. https://doi.org/10.3390/cells7110205
Chicago/Turabian StyleSegal, Tamar, Mali Salmon-Divon, and Gabi Gerlitz. 2018. "The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations" Cells 7, no. 11: 205. https://doi.org/10.3390/cells7110205
APA StyleSegal, T., Salmon-Divon, M., & Gerlitz, G. (2018). The Heterochromatin Landscape in Migrating Cells and the Importance of H3K27me3 for Associated Transcriptome Alterations. Cells, 7(11), 205. https://doi.org/10.3390/cells7110205