Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells

 , , , and
, , , and
Abstract
:1. Introduction
2. Elements of the Hh Signaling Pathway
2.1. Hh Proteins
2.2. PTCH
2.3. SMO
2.4. GLI
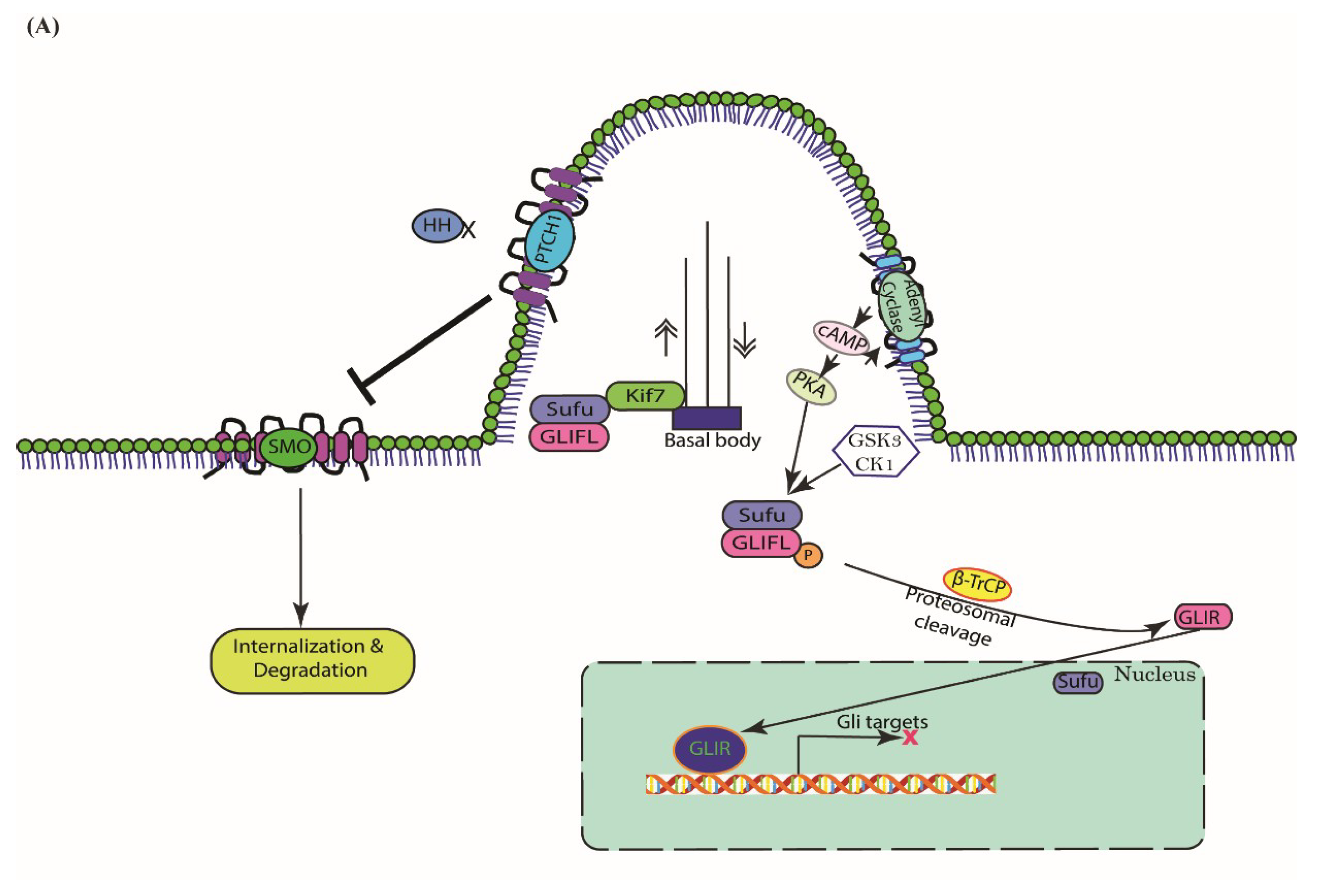
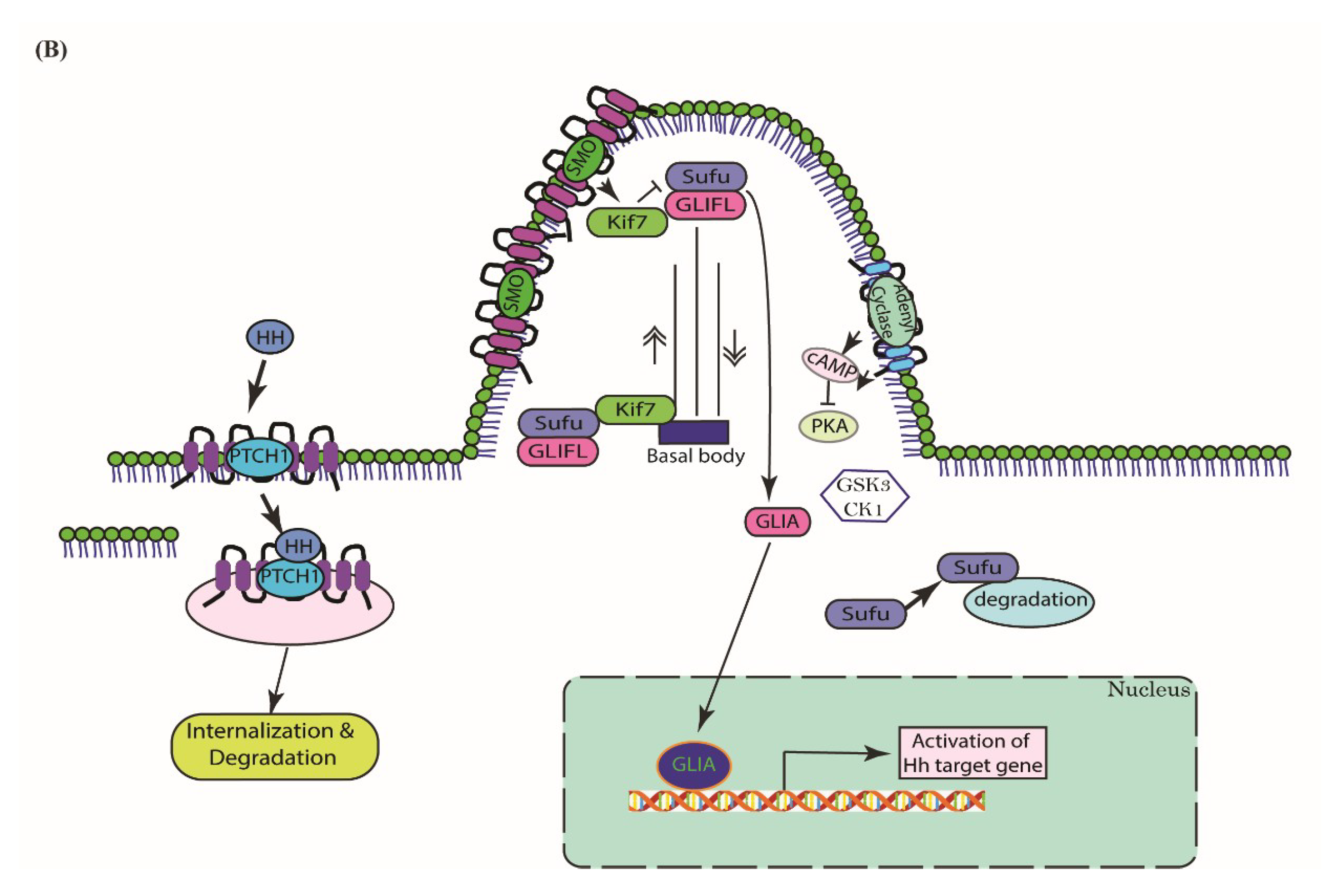
3. Cascades of Canonical Hh Signaling
4. Noncanonical Hedgehog Pathway
5. Types of Aberrant Activation of Hedgehog Signaling in Diseases and Cancers
5.1. Type I—Ligand Independent
5.2. Type II—Ligand-Dependent Autocrine/Juxtacrine Signaling
5.3. Type III—Ligand-Dependent Paracrine Signaling
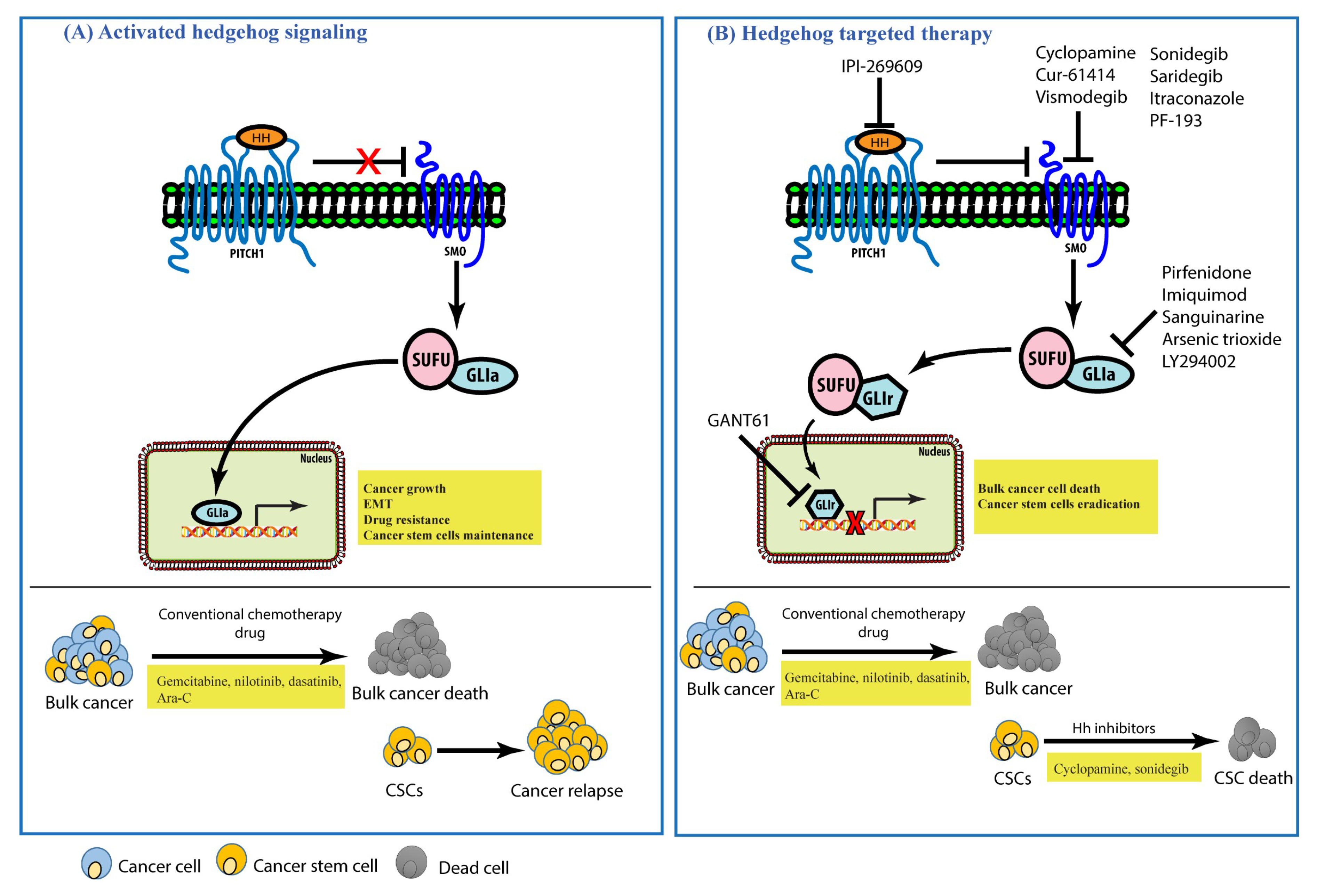
6. Hedgehog Signaling in Cancers and Its Inhibitors
6.1. Basal Cell Carcinoma (BCC)
6.2. Colon Cancer
6.3. Breast Cancer
6.4. Pancreatic Cancer
6.5. Medulloblastoma
6.6. Leukemia
7. Conclusions
Funding
Conflicts of Interest
References
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.W.; Chuang, P.T. Mechanism and evolution of cytosolic Hedgehog signal transduction. Development 2010, 137, 2079–2094. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Wang, Y.; Xie, J. The Hedgehog pathway: Role in cell differentiation, polarity and proliferation. Arch. Toxicol. 2015, 89, 179–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Haas, D.; Muenke, M. Abnormal sterol metabolism in holoprosencephaly. Am. J. Med. Genet. C Semin Med. Genet. 2010, 154C, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.E.; Heaney, S.J.; Lettice, L.A. Sonic hedgehog: Restricted expression and limb dysmorphologies. J. Anat. 2003, 202, 13–20. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol 2003, 53, 1–114. [Google Scholar] [PubMed]
- Muenke, M.; Beachy, P.A. Genetics of ventral forebrain development and holoprosencephaly. Curr. Opin. Genet. Dev. 2000, 10, 262–269. [Google Scholar] [CrossRef] [Green Version]
- Tostar, U.; Toftgard, R.; Zaphiropoulos, P.G.; Shimokawa, T. Reduction of human embryonal rhabdomyosarcoma tumor growth by inhibition of the hedgehog signaling pathway. Genes Cancer 2010, 1, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.M.; Sun, Z.J.; Bian, Z.; Fan, M.W.; Chen, Z. Epithelial expression of SHH signaling pathway in odontogenic tumors. Oral. Oncol. 2006, 42, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Hui, M.; Cazet, A.; Nair, R.; Watkins, D.N.; O’Toole, S.A.; Swarbrick, A. The Hedgehog signalling pathway in breast development, carcinogenesis and cancer therapy. Breast Cancer Res. 2013, 15, 203. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Singh, R.R.; Vega, F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am. J. Pathol. 2012, 180, 2–11. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Swarbrick, A.; Sutherland, R.L. The Hedgehog signalling pathway as a therapeutic target in early breast cancer development. Expert Opin. Ther. Targets 2009, 13, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Teglund, S.; Toftgard, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Feng, J.; Seidel, K.; Shi, S.; Klein, O.; Sharpe, P.; Chai, Y. Secretion of shh by a neurovascular bundle niche supports mesenchymal stem cell homeostasis in the adult mouse incisor. Cell Stem Cell 2014, 14, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, B.; Syn, W.K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.; Lee, J.; Guo, N.; Kim, J.; Lim, A.; Qu, L.; Mysorekar, I.U.; Beachy, P.A. Hedgehog/Wnt feedback supports regenerative proliferation of epithelial stem cells in bladder. Nature 2011, 472, 110–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubin, L.L.; de Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Zhang, Y.; Sun, B.; McMahon, A.P.; Wang, Y. Hedgehog Signaling: From Basic Biology to Cancer Therapy. Cell Chem. Biol. 2017, 24, 252–280. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 2008, 14, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Kawaguchi, H.; Kugimiya, F.; Ogasawara, T.; Kawamura, N.; Saito, T.; Ikeda, T.; Fujii, K.; Miyajima, T.; Kuramochi, A.; et al. Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Dev. Cell 2008, 14, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Wijgerde, M.; Ooms, M.; Hoogerbrugge, J.W.; Grootegoed, J.A. Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology 2005, 146, 3558–3566. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.H.; Whoriskey, W.; Capel, B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002, 16, 1433–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, P.J.; Guasti, L.; Laufer, E. Hedgehog signalling in endocrine development and disease. J. Endocrinol. 2008, 198, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, R.K.; Beachy, P.A. Novel lipid modifications of secreted protein signals. Annu. Rev. Biochem. 2004, 73, 891–923. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Young, K.E.; Beachy, P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science 1996, 274, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Buglino, J.A.; Resh, M.D. Hhat is a palmitoylacyltransferase with specificity for N-palmitoylation of Sonic Hedgehog. J. Biol. Chem. 2008, 283, 22076–22088. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Li, Y.J.; Kawakami, T.; Xu, S.M.; Chuang, P.T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004, 18, 641–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallet, A.; Ruel, L.; Staccini-Lavenant, L.; Therond, P.P. Cholesterol modification is necessary for controlled planar long-range activity of Hedgehog in Drosophila epithelia. Development 2006, 133, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, J.A.; Suber, L.M.; Zeng, X.; Robbins, D.J. Sonic Hedgehog as a mediator of long-range signaling. Bioessays 2002, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Weed, M.; Mundlos, S.; Olsen, B.R. The role of sonic hedgehog in vertebrate development. Matrix Biol. 1997, 16, 53–58. [Google Scholar] [CrossRef]
- Bellusci, S.; Furuta, Y.; Rush, M.G.; Henderson, R.; Winnier, G.; Hogan, B.L. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 1997, 124, 53–63. [Google Scholar] [PubMed]
- Litingtung, Y.; Lei, L.; Westphal, H.; Chiang, C. Sonic hedgehog is essential to foregut development. Nat. Genet. 1998, 20, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, Z.; Mo, R.; Hui, C.C.; Sharpe, P.T. The Shh signalling pathway in tooth development: Defects in Gli2 and Gli3 mutants. Development 1998, 125, 2803–2811. [Google Scholar] [PubMed]
- Seppala, M.; Fraser, G.J.; Birjandi, A.A.; Xavier, G.M.; Cobourne, M.T. Sonic Hedgehog Signaling and Development of the Dentition. J. Dev. Biol. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Schock, E.N.; Brugmann, S.A. Neural crest cells utilize primary cilia to regulate ventral forebrain morphogenesis via Hedgehog-dependent regulation of oriented cell division. Dev. Biol. 2017, 431, 168–178. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol. 1998, 8, 1058–1068. [Google Scholar] [CrossRef]
- Apelqvist, A.; Ahlgren, U.; Edlund, H. Sonic hedgehog directs specialised mesoderm differentiation in the intestine and pancreas. Curr. Biol. 1997, 7, 801–804. [Google Scholar] [CrossRef]
- Van den Brink, G.R.; Hardwick, J.C.; Nielsen, C.; Xu, C.; ten Kate, F.J.; Glickman, J.; van Deventer, S.J.; Roberts, D.J.; Peppelenbosch, M.P. Sonic hedgehog expression correlates with fundic gland differentiation in the adult gastrointestinal tract. Gut 2002, 51, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Aihara, E.; Kenny, S.; Yang, L.; Li, J.; Varro, A.; Montrose, M.H.; Shroyer, N.F.; Wang, T.C.; Shivdasani, R.A.; et al. Indian Hedgehog mediates gastrin-induced proliferation in stomach of adult mice. Gastroenterology 2014, 147, 655–666 e659. [Google Scholar] [CrossRef] [PubMed]
- Van den Brink, G.R.; Bleuming, S.A.; Hardwick, J.C.; Schepman, B.L.; Offerhaus, G.J.; Keller, J.J.; Nielsen, C.; Gaffield, W.; van Deventer, S.J.; Roberts, D.J.; et al. Indian Hedgehog is an antagonist of Wnt signaling in colonic epithelial cell differentiation. Nat. Genet. 2004, 36, 277–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, M.A.; Farrington, S.M.; Mohn, D.; Munday, J.R.; Baron, M.H. Indian hedgehog activates hematopoiesis and vasculogenesis and can respecify prospective neurectodermal cell fate in the mouse embryo. Development 2001, 128, 1717–1730. [Google Scholar] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Yang, K.; Chen, S.; Wang, J.; Du, G.; Fan, S.; Wei, L. Indian hedgehog contributes to human cartilage endplate degeneration. Eur. Spine J. 2015, 24, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Zuniga-Pflucker, J.C. An overview of the intrathymic intricacies of T cell development. J. Immunol. 2014, 192, 4017–4023. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Yanez, D.C.; Ross, S.; Lau, C.I.; Papaioannou, E.; Li, J.; Saldana, J.I.; Crompton, T. Gli3 in fetal thymic epithelial cells promotes thymocyte positive selection and differentiation by repression of Shh. Development 2018, 145, dev146910. [Google Scholar] [CrossRef] [PubMed]
- Outram, S.V.; Hager-Theodorides, A.L.; Shah, D.K.; Rowbotham, N.J.; Drakopoulou, E.; Ross, S.E.; Lanske, B.; Dessens, J.T.; Crompton, T. Indian hedgehog (Ihh) both promotes and restricts thymocyte differentiation. Blood 2009, 113, 2217–2228. [Google Scholar] [CrossRef] [PubMed]
- Solanki, A.; Lau, C.I.; Saldana, J.I.; Ross, S.; Crompton, T. The transcription factor Gli3 promotes B cell development in fetal liver through repression of Shh. J. Exp. Med. 2017, 214, 2041–2058. [Google Scholar] [CrossRef] [PubMed]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Lau, C.I.; Outram, S.V.; Saldana, J.I.; Furmanski, A.L.; Dessens, J.T.; Crompton, T. Regulation of murine normal and stress-induced erythropoiesis by Desert Hedgehog. Blood 2012, 119, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marigo, V.; Davey, R.A.; Zuo, Y.; Cunningham, J.M.; Tabin, C.J. Biochemical evidence that patched is the Hedgehog receptor. Nature 1996, 384, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Stone, D.M.; Hynes, M.; Armanini, M.; Swanson, T.A.; Gu, Q.; Johnson, R.L.; Scott, M.P.; Pennica, D.; Goddard, A.; Phillips, H.; et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature 1996, 384, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Chen, Y.; Jessell, T.M.; Struhl, G. A hedgehog-insensitive form of patched provides evidence for direct long-range morphogen activity of sonic hedgehog in the neural tube. Mol. Cell. 2001, 7, 1279–1291. [Google Scholar] [CrossRef]
- Ingham, P.W. How cholesterol modulates the signal. Curr. Biol. 2000, 10, R180–R183. [Google Scholar] [CrossRef]
- Carpenter, D.; Stone, D.M.; Brush, J.; Ryan, A.; Armanini, M.; Frantz, G.; Rosenthal, A.; de Sauvage, F.J. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc. Natl. Acad. Sci. USA 1998, 95, 13630–13634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gailani, M.R.; Stahle-Backdahl, M.; Leffell, D.J.; Glynn, M.; Zaphiropoulos, P.G.; Pressman, C.; Unden, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Smyth, I.; Narang, M.A.; Evans, T.; Heimann, C.; Nakamura, Y.; Chenevix-Trench, G.; Pietsch, T.; Wicking, C.; Wainwright, B.J. Isolation and characterization of human patched 2 (PTCH2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum. Mol. Genet. 1999, 8, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcedo, J.; Ayzenzon, M.; Von Ohlen, T.; Noll, M.; Hooper, J.E. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the hedgehog signal. Cell 1996, 86, 221–232. [Google Scholar] [CrossRef]
- Rana, R.; Carroll, C.E.; Lee, H.J.; Bao, J.; Marada, S.; Grace, C.R.; Guibao, C.D.; Ogden, S.K.; Zheng, J.J. Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat. Commun. 2013, 4, 2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murone, M.; Rosenthal, A.; de Sauvage, F.J. Hedgehog signal transduction: From flies to vertebrates. Exp. Cell Res. 1999, 253, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. In vivo evidence that Patched and Smoothened constitute distinct binding and transducing components of a Hedgehog receptor complex. Development 1998, 125, 4943–4948. [Google Scholar] [PubMed]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Tong, C.; Wang, B.; Luo, L.; Jiang, J. Hedgehog signalling activity of Smoothened requires phosphorylation by protein kinase A and casein kinase I. Nature 2004, 432, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Liu, C.Z.; Yang, J.T.; Swart, R.; Iannaccone, P.; Walterhouse, D. GLI activates transcription through a herpes simplex viral protein 16-like activation domain. J. Biol. Chem. 1998, 273, 3496–3501. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Hui, C.; Nakafuku, M.; Kondoh, H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development 1997, 124, 1313–1322. [Google Scholar] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, A.A.; Goode, D.K.; Amir, S.; Grzeschik, K.H. Evolution and functional diversification of the GLI family of transcription factors in vertebrates. Evol. Bioinform. Online 2009, 5, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Hynes, M.; Stone, D.M.; Dowd, M.; Pitts-Meek, S.; Goddard, A.; Gurney, A.; Rosenthal, A. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron 1997, 19, 15–26. [Google Scholar] [CrossRef]
- Santoni, M.; Burattini, L.; Nabissi, M.; Morelli, M.B.; Berardi, R.; Santoni, G.; Cascinu, S. Essential role of Gli proteins in glioblastoma multiforme. Curr. Protein Pept. Sci. 2013, 14, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Persson, M.; Stamataki, D.; te Welscher, P.; Andersson, E.; Bose, J.; Ruther, U.; Ericson, J.; Briscoe, J. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 2002, 16, 2865–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aza-Blanc, P.; Lin, H.Y.; Ruiz i Altaba, A.; Kornberg, T.B. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 2000, 127, 4293–4301. [Google Scholar] [PubMed]
- Dai, P.; Akimaru, H.; Tanaka, Y.; Maekawa, T.; Nakafuku, M.; Ishii, S. Sonic Hedgehog-induced activation of the Gli1 promoter is mediated by GLI3. J. Biol. Chem. 1999, 274, 8143–8152. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [PubMed]
- Denef, N.; Neubuser, D.; Perez, L.; Cohen, S.M. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell 2000, 102, 521–531. [Google Scholar] [CrossRef]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Ruiz, I.A.A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol 2014, 33, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Mastronardi, F.G.; Dimitroulakos, J.; Kamel-Reid, S.; Manoukian, A.S. Co-localization of patched and activated sonic hedgehog to lysosomes in neurons. Neuroreport 2000, 11, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Humke, E.W.; Dorn, K.V.; Milenkovic, L.; Scott, M.P.; Rohatgi, R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010, 24, 670–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liem, K.F., Jr.; He, M.; Ocbina, P.J.; Anderson, K.V. Mouse Kif7/Costal2 is a cilia-associated protein that regulates Sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 13377–13382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, H.O.; Zhang, X.; Ribeiro, A.; Mo, R.; Makino, S.; Puviindran, V.; Law, K.K.; Briscoe, J.; Hui, C.C. The kinesin protein Kif7 is a critical regulator of Gli transcription factors in mammalian hedgehog signaling. Sci. Signal. 2009, 2, ra29. [Google Scholar] [CrossRef] [PubMed]
- Endoh-Yamagami, S.; Evangelista, M.; Wilson, D.; Wen, X.; Theunissen, J.W.; Phamluong, K.; Davis, M.; Scales, S.J.; Solloway, M.J.; de Sauvage, F.J.; et al. The mammalian Cos2 homolog Kif7 plays an essential role in modulating Hh signal transduction during development. Curr. Biol. 2009, 19, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Kicheva, A.; Ribeiro, A.; Blassberg, R.; Page, K.M.; Barnes, C.P.; Briscoe, J. Ptch1 and Gli regulate Shh signalling dynamics via multiple mechanisms. Nat. Commun. 2015, 6, 6709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical Hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Regan, J.L.; Schumacher, D.; Staudte, S.; Steffen, A.; Haybaeck, J.; Keilholz, U.; Schweiger, C.; Golob-Schwarzl, N.; Mumberg, D.; Henderson, D.; et al. Non-Canonical Hedgehog Signaling Is a Positive Regulator of the WNT Pathway and Is Required for the Survival of Colon Cancer Stem Cells. Cell Rep. 2017, 21, 2813–2828. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, E.; Matevossian, A.; Resh, M.D. Hedgehog acyltransferase as a target in pancreatic ductal adenocarcinoma. Oncogene 2015, 34, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajurkar, M.; De Jesus-Monge, W.E.; Driscoll, D.R.; Appleman, V.A.; Huang, H.; Cotton, J.L.; Klimstra, D.S.; Zhu, L.J.; Simin, K.; Xu, L.; et al. The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. USA 2012, 109, E1038–E1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Yang, P.; Ma, L. Kinase activity-independent regulation of cyclin pathway by GRK2 is essential for zebrafish early development. Proc. Natl. Acad. Sci. USA 2009, 106, 10183–10188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link Hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijlsma, M.F.; Borensztajn, K.S.; Roelink, H.; Peppelenbosch, M.P.; Spek, C.A. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell Signal. 2007, 19, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Peppelenbosch, M.P.; Spek, C.A.; Roelink, H. Leukotriene synthesis is required for hedgehog-dependent neurite projection in neuralized embryoid bodies but not for motor neuron differentiation. Stem Cells 2008, 26, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Yam, P.T.; Langlois, S.D.; Morin, S.; Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron 2009, 62, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Lo Muzio, L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet J. Rare Dis. 2008, 3, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, L.V.; Milenkovic, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgard, R.; Unden, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.W.; Dalton, J.; Kocak, M.; Nicholson, S.L.; Fraga, C.; Neale, G.; Kenney, A.M.; Brat, D.J.; Perry, A.; Yong, W.H.; et al. Medulloblastoma: Clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011, 121, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- McGarvey, T.W.; Maruta, Y.; Tomaszewski, J.E.; Linnenbach, A.J.; Malkowicz, S.B. PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene 1998, 17, 1167–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maesawa, C.; Tamura, G.; Iwaya, T.; Ogasawara, S.; Ishida, K.; Sato, N.; Nishizuka, S.; Suzuki, Y.; Ikeda, K.; Aoki, K.; et al. Mutations in the human homologue of the Drosophila patched gene in esophageal squamous cell carcinoma. Genes Chromosomes Cancer 1998, 21, 276–279. [Google Scholar] [CrossRef]
- Vorechovsky, I.; Unden, A.B.; Sandstedt, B.; Toftgard, R.; Stahle-Backdahl, M. Trichoepitheliomas contain somatic mutations in the overexpressed PTCH gene: Support for a gatekeeper mechanism in skin tumorigenesis. Cancer Res. 1997, 57, 4677–4681. [Google Scholar] [PubMed]
- Adolphe, C.; Hetherington, R.; Ellis, T.; Wainwright, B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006, 66, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Aszterbaum, M.; Epstein, J.; Oro, A.; Douglas, V.; LeBoit, P.E.; Scott, M.P.; Epstein, E.H., Jr. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat. Med. 1999, 5, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Svard, J.; Heby-Henricson, K.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergstrom, A.; Ericson, J.; Toftgard, R.; Teglund, S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell. 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1086. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Datta, M.W. Sonic Hedgehog signaling in advanced prostate cancer. Cell. Mol. Life Sci. 2006, 63, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; He, J.; Zhang, X.; Bian, Y.; Yang, L.; Xie, G.; Zhang, K.; Tang, W.; Stelter, A.A.; Wang, Q.; et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 2006, 27, 1334–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Szkandera, J.; Kiesslich, T.; Haybaeck, J.; Gerger, A.; Pichler, M. Hedgehog signaling pathway in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 1179–1196. [Google Scholar] [CrossRef] [PubMed]
- Dahmane, N.; Sanchez, P.; Gitton, Y.; Palma, V.; Sun, T.; Beyna, M.; Weiner, H.; Ruiz i Altaba, A. The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development 2001, 128, 5201–5212. [Google Scholar] [PubMed]
- O’Reilly, K.E.; de Miera, E.V.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals (Basel) 2013, 6, 1429–1450. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; DuPree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Sheng, T.; Zhang, Y.; Zhang, X.; He, J.; Huang, S.; Chen, K.; Sultz, J.; Adegboyega, P.A.; Zhang, H.; et al. Hedgehog signaling is activated in subsets of esophageal cancers. Int. J. Cancer 2006, 118, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Blotta, S.; Jakubikova, J.; Calimeri, T.; Roccaro, A.M.; Amodio, N.; Azab, A.K.; Foresta, U.; Mitsiades, C.S.; Rossi, M.; Todoerti, K.; et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012, 120, 5002–5013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, P.; Wang, F.; Yang, J.; Yang, Z.; Qin, H. The relationship between early embryo development and tumourigenesis. J. Cell. Mol. Med. 2010, 14, 2697–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. Hedgehog-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, X.; Gao, S.; Zhang, C.; Qu, C.; Wang, P.; Liu, L. Ski modulate the characteristics of pancreatic cancer stem cells via regulating sonic hedgehog signaling pathway. Tumour Biol. 2016. [Google Scholar] [CrossRef]
- Quint, K.; Tonigold, M.; Di Fazio, P.; Montalbano, R.; Lingelbach, S.; Ruckert, F.; Alinger, B.; Ocker, M.; Neureiter, D. Pancreatic cancer cells surviving gemcitabine treatment express markers of stem cell differentiation and epithelial-mesenchymal transition. Int. J. Oncol. 2012, 41, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Kiesslich, T.; Mayr, C.; Wachter, J.; Bach, D.; Fuereder, J.; Wagner, A.; Alinger, B.; Pichler, M.; Di Fazio, P.; Ocker, M.; et al. Activated hedgehog pathway is a potential target for pharmacological intervention in biliary tract cancer. Mol. Cell. Biochem. 2014, 396, 257–268. [Google Scholar] [CrossRef] [PubMed]
- P, D.I.F.; Montalbano, R.; Quint, K.; Alinger, B.; Kemmerling, R.; Kiesslich, T.; Ocker, M.; Neureiter, D. The pan-deacetylase inhibitor panobinostat modulates the expression of epithelial-mesenchymal transition markers in hepatocellular carcinoma models. Oncol. Lett. 2013, 5, 127–134. [Google Scholar] [CrossRef]
- Chen, W.; Tang, T.; Eastham-Anderson, J.; Dunlap, D.; Alicke, B.; Nannini, M.; Gould, S.; Yauch, R.; Modrusan, Z.; DuPree, K.J.; et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9589–9594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell. 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell. 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results From Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Shimizu, Y.; Nakashima, K.; Kondo, S.; Ogawa, K.; Sasaki, S.; Matsui, H. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur. J. Pharmacol. 2014, 723, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.R.; Zhao, H.; Zhang, X.S.; Lang, H.; Yu, K. Novel-smoothened inhibitors for therapeutic targeting of naive and drug-resistant hedgehog pathway-driven cancers. Acta Pharmacol. Sin. 2018. [Google Scholar] [CrossRef]
- Colmont, C.S.; Benketah, A.; Reed, S.H.; Hawk, N.V.; Telford, W.G.; Ohyama, M.; Udey, M.C.; Yee, C.L.; Vogel, J.C.; Patel, G.K. CD200-expressing human basal cell carcinoma cells initiate tumor growth. Proc. Natl. Acad. Sci. USA 2013, 110, 1434–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, V.; Ruiz i Altaba, A. Hedgehog-GLI signaling regulates the behavior of cells with stem cell properties in the developing neocortex. Development 2004, 131, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, D.; Liu, H.; Su, G.H.; Zhang, X.; Chin-Sinex, H.; Hanenberg, H.; Mendonca, M.S.; Shannon, H.E.; Chiorean, E.G.; Xie, J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol. Cancer Ther. 2013, 12, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.T.; Zhuan-Sun, Y.X.; Zhuang, Y.Y.; Wei, S.L.; Tang, J.; Chen, W.B.; Zhang, S.N. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-H.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sulforaphane regulates self-renewal of pancreatic cancer stem cells through the modulation of Sonic hedgehog–GLI pathway. Mol. Cell. Biochem. 2013, 373, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yu, W.; Shrivastava, A.; Alemi, F.; Lankachandra, K.; Srivastava, R.K.; Shankar, S. Sanguinarine inhibits pancreatic cancer stem cell characteristics by inducing oxidative stress and suppressing sonic hedgehog-Gli-Nanog pathway. Carcinogenesis 2017, 38, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chen, X.; Wang, P.; Gao, S.; Qu, C.; Liu, L. Effects of baicalein on pancreatic cancer stem cells via modulation of sonic Hedgehog pathway. Acta Biochim. Biophys. Sin. 2018, 50, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; An, Y.; Wie, J.S.; Ji, Z.L.; Lu, Z.P.; Wu, J.L.; Jiang, K.R.; Chen, P.; Xu, Z.K.; Miao, Y. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med. Wkly. 2011, 141, w13208. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Ferretti, E.; Miele, E.; De Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; Di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, T.A.; Fogarty, M.P.; Markant, S.L.; McLendon, R.E.; Wei, Z.; Ellison, D.W.; Febbo, P.G.; Wechsler-Reya, R.J. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 2009, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Irvine, D.A.; Zhang, B.; Allan, E.K.; Holyoake, T.L.; Dorsch, M.; Manley, P.W.; Bhatia, R.; Copland, M. Combination of the hedgehog pathway inhibitor LDE225 and nilotinib eliminates chronic myeloid leukemia stem and progenitor cells. Blood 2009, 114, 1428. [Google Scholar]
- Queiroz, K.C.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, S.; Minami, Y.; Miyata, Y.; Mizutani, Y.; Goto, H.; Kawamoto, S.; Yakushijin, K.; Kurata, K.; Matsuoka, H.; Minami, H. NANOG expression as a responsive biomarker during treatment with Hedgehog signal inhibitor in acute myeloid leukemia. Int. J. Mol. Sci. 2017, 18, 486. [Google Scholar] [CrossRef] [PubMed]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minami, Y.; Hayakawa, F.; Kiyoi, H.; Sadarangani, A.; Jamieson, C.H.; Naoe, T. Treatment with hedgehog inhibitor, PF-04449913, attenuates leukemia-initiation potential in acute myeloid leukemia cells. Blood 2013, 122, 1649. [Google Scholar]
- Ji, Z.; Mei, F.C.; Johnson, B.H.; Thompson, E.B.; Cheng, X. Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J. Biol. Chem. 2007, 282, 37370–37377. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Badura, S.; Ruthardt, M.; Rieger, M.A.; Ottmann, O.G. Modulation of Leukemic Stem Cell Self-Renewal and Cell Fate Decisions by Inhibition of Hedgehog Signalling in Human Acute Lymphoblastic Leukemia (ALL). Blood 2012, 120, 2578. [Google Scholar]
- Lin, T.L.; Wang, Q.H.; Brown, P.; Peacock, C.; Merchant, A.A.; Brennan, S.; Jones, E.; McGovern, K.; Watkins, D.N.; Sakamoto, K.M. Self-renewal of acute lymphocytic leukemia cells is limited by the Hedgehog pathway inhibitors cyclopamine and IPI-926. PLoS ONE 2010, 5, e15262. [Google Scholar] [CrossRef] [PubMed]
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Dreier, J.; Cheng, P.F.; Nageli, M.; Lehmann, H.; Felderer, L.; Frew, I.J.; Matsushita, S.; Levesque, M.P.; Dummer, R. Hedgehog pathway inhibitors promote adaptive immune responses in basal cell carcinoma. Clin. Cancer Res. 2015, 21, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Graves, S.; Meng, F.; Mandal, M.; Mashayekhi, M.; Aifantis, I. Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat. Immunol. 2006, 7, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Furmanski, A.L.; Barbarulo, A.; Solanki, A.; Lau, C.I.; Sahni, H.; Saldana, J.I.; D’Acquisto, F.; Crompton, T. The transcriptional activator Gli2 modulates T-cell receptor signalling through attenuation of AP-1 and NFkappaB activity. J. Cell. Sci. 2015, 128, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Hager-Theodorides, A.L.; Cebecauer, M.; Shah, D.K.; Drakopoulou, E.; Dyson, J.; Outram, S.V.; Crompton, T. Activation of the Hedgehog signaling pathway in T-lineage cells inhibits TCR repertoire selection in the thymus and peripheral T-cell activation. Blood 2007, 109, 3757–3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jiang, J.; Wang, Z.; Zhang, J.; Xiao, M.; Wang, C.; Lu, Y.; Qin, Z. Endogenous interleukin-4 promotes tumor development by increasing tumor cell resistance to apoptosis. Cancer Res. 2008, 68, 8687–8694. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Heidenreich, R.; Braumuller, H.; Wolburg, H.; Weidemann, S.; Mocikat, R.; Rocken, M. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood 2009, 113, 3494–3502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furmanski, A.L.; Saldana, J.I.; Ono, M.; Sahni, H.; Paschalidis, N.; D’Acquisto, F.; Crompton, T. Tissue-derived hedgehog proteins modulate Th differentiation and disease. J. Immunol. 2013, 190, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Rowbotham, N.J.; Furmanski, A.L.; Hager-Theodorides, A.L.; Ross, S.E.; Drakopoulou, E.; Koufaris, C.; Outram, S.V.; Crompton, T. Repression of hedgehog signal transduction in T-lineage cells increases TCR-induced activation and proliferation. Cell Cycle 2008, 7, 904–908. [Google Scholar] [CrossRef] [PubMed]
- Tabs, S.; Avci, O. Induction of the differentiation and apoptosis of tumor cells in vivo with efficiency and selectivity. Eur. J. Dermatol. 2004, 14, 96–102. [Google Scholar] [PubMed]
- Athar, M.; Li, C.; Tang, X.; Chi, S.; Zhang, X.; Kim, A.L.; Tyring, S.K.; Kopelovich, L.; Hebert, J.; Epstein, E.H., Jr.; et al. Inhibition of smoothened signaling prevents ultraviolet B-induced basal cell carcinomas through regulation of Fas expression and apoptosis. Cancer Res. 2004, 64, 7545–7552. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Chang, A.L.; Oro, A.E. Hedgehog pathway inhibition and the race against tumor evolution. J. Cell. Biol. 2012, 199, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, H.; Olivero, C.; Patel, G.K. Identification of Human Cutaneous Basal Cell Carcinoma Cancer Stem Cells. Methods Mol. Biol. 2018. [Google Scholar] [CrossRef]
- Li, T.; Liao, X.; Lochhead, P.; Morikawa, T.; Yamauchi, M.; Nishihara, R.; Inamura, K.; Kim, S.A.; Mima, K.; Sukawa, Y.; et al. SMO expression in colorectal cancer: Associations with clinical, pathological, and molecular features. Ann. Surg. Oncol. 2014, 21, 4164–4173. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, K.; Shimada, M.; Miyamoto, H.; Higashijima, J.; Miyatani, T.; Nishioka, M.; Kurita, N.; Iwata, T.; Uehara, H. Sonic hedgehog relates to colorectal carcinogenesis. J. Gastroenterol. 2009, 44, 1113–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, S.S.; Wei, G.J.; Deng, Z.M.; Hu, Y. Dysregulation of hedgehog signaling pathway related components in the evolution of colonic carcinogenesis. Int. J. Clin. Exp. Med. 2015, 8, 21379–21385. [Google Scholar] [PubMed]
- Bian, Y.H.; Huang, S.H.; Yang, L.; Ma, X.L.; Xie, J.W.; Zhang, H.W. Sonic hedgehog-Gli1 pathway in colorectal adenocarcinomas. World J. Gastroenterol. 2007, 13, 1659–1665. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Shimoji, T.; Takifuji, K.; Hotta, T.; Yokoyama, S.; Matsuda, K.; Higashiguchi, T.; Tominaga, T.; Nasu, T.; Tamura, K.; et al. Identification of the molecular mechanisms for dedifferentiation at the invasion front of colorectal cancer by a gene expression analysis. Clin. Cancer Res. 2008, 14, 7215–7222. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Xu, X.F.; Xu, L.; Niu, P.Q.; Wang, F.; Hu, G.Y.; Wang, X.P.; Guo, C.Y. Cyclopamine blocked the growth of colorectal cancer SW116 cells by modulating some target genes of Gli1 in vitro. Hepatogastroenterology 2011, 58, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Qualtrough, D.; Rees, P.; Speight, B.; Williams, A.C.; Paraskeva, C. The Hedgehog Inhibitor Cyclopamine Reduces beta-Catenin-Tcf Transcriptional Activity, Induces E-Cadherin Expression, and Reduces Invasion in Colorectal Cancer Cells. Cancers (Basel) 2015, 7, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, H.; Wu, M.; Wang, Q.; Luo, L.; Ma, H.; Zhang, X.; He, S. Discovery of a potent hedgehog pathway inhibitor capable of activating caspase8-dependent apoptosis. J. Pharmacol. Sci. 2018, 137, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Li, Z.Y.; Liu, W.P.; Zhao, M.R. Crosstalk between Wnt/beta-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol. Ther. 2015, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Yu, K.; Zhang, L.; Li, Y.; Li, Q.; Yang, Z.; Shen, T.; Duan, L.; Xiong, W.; Wang, W. Synergistic inhibition of colon carcinoma cell growth by Hedgehog-Gli1 inhibitor arsenic trioxide and phosphoinositide 3-kinase inhibitor LY294002. OncoTargets Ther. 2015, 8, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Batsaikhan, B.E.; Yoshikawa, K.; Kurita, N.; Iwata, T.; Takasu, C.; Kashihara, H.; Shimada, M. Cyclopamine decreased the expression of Sonic Hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014, 34, 6339–6344. [Google Scholar] [PubMed]
- Didiasova, M.; Schaefer, L.; Wygrecka, M. Targeting GLI Transcription Factors in Cancer. Molecules 2018, 23, 1003. [Google Scholar] [CrossRef] [PubMed]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Hunsberger, S.; Yang, S.X. Expression of Gli1 in the hedgehog signaling pathway and breast cancer recurrence. Chin. J. Cancer Res. 2012, 24, 257–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of Hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, J.; Kanomata, N.; Koike, Y.; Ohta, Y.; Saitoh, W.; Kishino, E. Comprehensive immunohistochemical analyses on expression levels of hedgehog signaling molecules in breast cancers. Breast Cancer 2018, 25, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Borrego, M.; Jimenez, B.; Antolin, S.; Garcia-Saenz, J.A.; Corral, J.; Jerez, Y.; Trigo, J.; Urruticoechea, A.; Colom, H.; Gonzalo, N.; et al. A phase Ib study of sonidegib (LDE225), an oral small molecule inhibitor of smoothened or Hedgehog pathway, in combination with docetaxel in triple negative advanced breast cancer patients: GEICAM/2012-12 (EDALINE) study. Investig. New Drugs 2018. [Google Scholar] [CrossRef]
- Wolf, I.; Bose, S.; Desmond, J.C.; Lin, B.T.; Williamson, E.A.; Karlan, B.Y.; Koeffler, H.P. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer. Breast Cancer Res. Treat. 2007, 105, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef] [PubMed]
- Salazar, L.G.; Lu, H.; Reichow, J.L.; Childs, J.S.; Coveler, A.L.; Higgins, D.M.; Waisman, J.; Allison, K.H.; Dang, Y.; Disis, M.L. Topical Imiquimod Plus Nab-paclitaxel for Breast Cancer Cutaneous Metastases: A Phase 2 Clinical Trial. JAMA Oncol. 2017, 3, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgard, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1)expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ercan, G.; Karlitepe, A.; Ozpolat, B. Pancreatic cancer stem cells and therapeutic approaches. Anticancer Res. 2017, 37, 2761–2775. [Google Scholar] [PubMed]
- Lau, J.; Kawahira, H.; Hebrok, M. Hedgehog signaling in pancreas development and disease. Cell. Mol. Life Sci. 2006, 63, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Sato, N.; Dhara, S.; Chang, R.; Hustinx, S.R.; Abe, T.; Maitra, A.; Goggins, M. Aberrant methylation of the Human Hedgehog interacting protein (HHIP) gene in pancreatic neoplasms. Cancer Biol. Ther. 2005, 4, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Habbe, N.; Dhara, S.; Bisht, S.; Alvarez, H.; Fendrich, V.; Beaty, R.; Mullendore, M.; Karikari, C.; Oullette, M.M. Hedgehog inhibition prolongs survival in a genetically engineered mouse model of pancreatic cancer. Gut 2008, 57, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kelley, R.K.; Lewis, S.; Chang, W.-C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V. A phase I study of FOLFIRINOX plus IPI-926, a hedgehog pathway inhibitor, for advanced pancreatic adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Balis, U.G.; Craig, R. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, T.; Waha, A.; Koch, A.; Kraus, J.; Albrecht, S.; Tonn, J.; Sorensen, N.; Berthold, F.; Henk, B.; Schmandt, N.; et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997, 57, 2085–2088. [Google Scholar] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell. 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Hegde, G.V.; Peterson, K.J.; Emanuel, K.; Mittal, A.K.; Joshi, A.D.; Dickinson, J.D.; Kollessery, G.J.; Bociek, R.G.; Bierman, P.; Vose, J.M. Hedgehog-induced survival of B-cell chronic lymphocytic leukemia cells in a stromal cell microenvironment: A potential new therapeutic target. Mol. Cancer Res. 2008, 6, 1928–1936. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Zhu, H.; Zhu, C.; Liu, T.; Meng, W. Activation of the Hedgehog pathway in chronic myelogeneous leukemia patients. J. Exp. Clin. Cancer Res. 2011, 30, 8. [Google Scholar] [CrossRef] [PubMed]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef] [PubMed]
- Ochs, M.; Farrar, J.; Considine, M.; Ries, R.E.; Trevino, L.R.; Alonzo, T.A.; Guidry-Auvil, J.; Davidsen, T.M.; Gesuwan, P.; Muzny, D.M. Genome wide promoter methylation patterns predict AML subtype outcomes and identify novel pathways characterizing diagnostic and relapsed disease in children. Blood 2012, 120, 1287. [Google Scholar]
- Campbell, V.; Tholouli, E.; Quigley, M.T.; Keilty, J.; Kelly, P.; McGovern, K.; Read, M.; Kutok, J.L.; Byers, R. Evidence that activated hedgehog signaling predicts for poor clinical outcome in acute myeloid leukemia. Blood 2012, 120, 1441. [Google Scholar]
- Burns, M.A.; Liao, Z.W.; Yamagata, N.; Pouliot, G.P.; Stevenson, K.E.; Neuberg, D.S.; Thorner, A.R.; Ducar, M.; Silverman, E.A.; Hunger, S.P. Hedgehog pathway mutations drive oncogenic transformation in high-risk T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Dagklis, A.; Demeyer, S.; De Bie, J.; Radaelli, E.; Pauwels, D.; Degryse, S.; Gielen, O.; Vicente, C.; Vandepoel, R.; Geerdens, E.; et al. Hedgehog pathway activation in T-cell acute lymphoblastic leukemia predicts response to SMO and GLI1 inhibitors. Blood 2016, 128, 2642–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagklis, A.; Pauwels, D.; Lahortiga, I.; Geerdens, E.; Bittoun, E.; Cauwelier, B.; Tousseyn, T.; Uyttebroeck, A.; Maertens, J.; Verhoef, G.; et al. Hedgehog pathway mutations in T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, e102–105. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.; et al. Hedgehog signaling is dispensable for adult hematopoietic stem cell function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type | Characteristic | Cancer Type |
|---|---|---|
| Type I | Ligand independent | Basal cell carcinoma Medulloblastoma (MB) Pediatric brain tumor & Rhabdomyosarcoma |
| Type II | Ligand-dependent autocrine/juxtacrine signaling | Colorectal Prostate Liver Breast Ovarian Brain Melanoma |
| Type III | Ligand-dependent paracrine signaling | Pancreatic Leukemia (reverse paracrine) |
| Tumor Type | CSC Marker | Hh Inhibitors | Combination Therapy | References |
|---|---|---|---|---|
| Basal cell carcinoma (BCC) | CD200+CD45− | Smo antagonist | CD200+ neutralizing antibody | [164] |
| Colon cancer | CD133+ | PTCH1 inhibitor (RU-SKI 43), Cyclopamine | [97,128] | |
| Breast cancer | Lin−CD44+CD24− | Cyclopamine | [144,165] | |
| Pancreatic cancer | CD44+CD24+ESA+ | Sulforaphane, Baicalein, Sangunarine, GANT61 | Gemtacibine and cyclopamine, cyclopamine derivates (CyT) +2gy irradiation | [166,167,168,169,170,171,172] |
| Medulloblastoma | CD15+, Sox2+ | KAAD-cyclopamine | [173,174,175] | |
| Chronic myelogenous leukemia (CML) | Lin−Sca1+cKit+CD34+ | Sonidegib, cyclopamine, PF-04449913 | Sonidegib and nilotinib, cyclopamine and nilotinib, PF-04449913 and dasatinib | [176,177,178,179] |
| Acute myelogenous leukemia (AML) | PF-04449913, cyclopamine, PF-913 | Cyclopamine and Ara-C | [180,181,182] | |
| Acute lymphoblastic leukemia (ALL) | Cyclopamine, IPI-926, KAAD-cyclopamine, SANT1 | [183,184,185] |
| Compound | Target | Conditions | Phase | NCT Number | Combination Drug | Locations | Status |
|---|---|---|---|---|---|---|---|
| GDC-0449 (Vismodegib/Erivedge) | SMO | Basal cell carcinoma | Early Phase 1 | NCT01631331 | - | US | Completed |
| Phase 1 | NCT02639117 | - | US | Completed | |||
| Phase 2 | NCT01543581 | - | - | Completed | |||
| NCT00833417 | - | US | Completed | ||||
| NCT01201915 | - | US | Completed | ||||
| NCT03035188 | - | Germany | Recruiting | ||||
| NCT02667574 | - | France | Active | ||||
| NCT01815840 | - | US | Completed | ||||
| NCT01367665 | - | Australia, Argentina | Completed | ||||
| NCT01898598 | - | US | Terminated | ||||
| NCT02067104 | - | US | Active | ||||
| NCT01700049 | - | US | Active | ||||
| NCT01835626 | - | US | Recruiting | ||||
| NCT01556009 | - | US | Completed | ||||
| NCT02956889 | - | Italy | Recruiting | ||||
| NCT00959647 | FOLFOX, FOLFIRI, Bevacizumab | US | Completed | ||||
| Phase 4 | NCT02436408 | - | US | Recruiting | |||
| Phase 1, 2 | NCT02690948 | Pembrolizumab | US | Active | |||
| - | NCT01160250 | - | US | Approved | |||
| NCT02371967 | - | Sweden | Active | ||||
| NCT02781389 | - | Germany | Active | ||||
| NCT02674009 | - | Germany | Active | ||||
| NCT02438644 | - | - | Completed | ||||
| Colon cancer | Phase 2 | NCT00636610 | Bevacizumab, Modified FOLFOX, FOLFIRI | - | Completed | ||
| NCT00959647 | FOLFOX, FOLFIRI, bevacizumab | US | Completed | ||||
| Breast cancer | Phase 2 | NCT02694224 | Paclitaxel, Epirubicin, Cyclophosphamide | Spain | Recruiting | ||
| Pancreatic cancer | Early Phase 1 | NCT01713218 | gemcitabine | Belgium | Unknown | ||
| Phase 1 | NCT00878163 | erlotinib hydrochloride, gemcitabine | US | Active | |||
| NCT01537107 | sirolimus | US | Suspended | ||||
| Phase 2 | NCT01088815 | Gemcitabine, nab-Paclitaxel | US | Unknown | |||
| NCT01195415 | Gemcitabine Hydrochloride | US | Completed | ||||
| Phase 1, 2 | NCT01064622 | gemcitabine hydrochloride | US | Completed | |||
| Medullo blastoma | Phase 1 | NCT00822458 | - | US | Completed | ||
| Phase 2 | NCT00939484 | - | US | Completed | |||
| NCT01239316 | - | US | Completed | ||||
| Phase 1, 2 | NCT01601184 | Temozolomide | France | Recruiting | |||
| Leukemia | Phase 2 | NCT01880437 | cytarabine | US | Terminated | ||
| NCT01944943 | - | France | Terminated | ||||
| NCT02073838 | Ribavirin, Decitabine | US | Completed | ||||
| LDE225 (Erismodegib/Sonidegib/Odomzo) | SMO | Basal cell carcinoma | Phase 1 | NCT01208831 | - | Hong Kong, Japan, Taiwan | Completed |
| NCT00880308 | - | US, Spain | Completed | ||||
| Phase 2 | NCT01327053 | - | US | Active | |||
| NCT02303041 | Buparlisib | US | Completed | ||||
| NCT01033019 | - | Austria, Australia | Terminated | ||||
| NCT03534947 | Imiquimod | Australia | Not yet | ||||
| NCT00961896 | - | Austria, Switzerland | Completed | ||||
| NCT01350115 | - | Austria, Belgium, Canada | Completed | ||||
| Phase 2, 3 | NCT03070691 | - | Belgium, Canada | Withdrawn | |||
| - | NCT01529450 | - | US | Completed | |||
| Breast cancer | Phase 1 | NCT02027376 | Docetaxel | Spain | Unknown | ||
| Phase 2 | NCT01757327 | - | - | Withdrawn | |||
| Pancreatic cancer | Early Phase 1 | NCT01694589 | - | US | Withdrawn | ||
| Phase 1 | NCT01487785 | gemcitabine | US | Completed | |||
| NCT01485744 | Fluorouracil, Leucovorin, Oxaliplatin, Irinotecan | US | Active | ||||
| Phase 1, 2 | NCT02358161 | nab paclitaxel | Netherland | Unknown | |||
| - | NCT01911416 | - | US | Withdrawn | |||
| Medullo blastoma | Phase 1 | NCT01208831 | - | Hong Kong, Japan, Taiwan | Completed | ||
| NCT00880308 | - | Spain, US | Completed | ||||
| Phase 2 | NCT01708174 | - | US | Completed | |||
| Phase 1, 2 | NCT01125800 | - | US | Completed | |||
| Leukemia | Phase 1 | NCT01456676 | nilotinib | Canada, France, Germany | Completed | ||
| NCT02129101 | Azacitidine, Decitabine | US | Active | ||||
| Phase 2 | NCT01826214 | - | Australia, US | Completed | |||
| IPI-926 (Saridegib) | SMO | Basal cell carcinoma | - | NCT01609179 | - | US | Completed |
| Pancreatic cancer | Phase 1 | NCT01383538 | 5-fluorouracil, Leucovorin, Irinotecan, Oxaliplatin | US | Completed | ||
| Phase 1, 2 | NCT01130142 | gemcitabine | US | Completed | |||
| PF-04449913 (Glasdegib) | SMO | Leukemia | Phase 1 | NCT01546038 | ARA-C, Decitabine, Daunorubicin, Cytarabine | US | Active |
| NCT02038777 | ARA-C, Daunorubicin, Cytarabine, Azacitidine | Japan | Recruiting | ||||
| NCT02367456 | Azacitidine | US | Recruiting | ||||
| NCT00953758 | - | US | Completed | ||||
| Phase 2 | NCT01841333 | - | US | Recruiting | |||
| NCT01842646 | - | US | Active | ||||
| NCT03390296 | Avelumab, azacitidine, utomilumab | US | Recruiting | ||||
| Phase 3 | NCT03416179 | - | US | Recruiting | |||
| BMS-833923 | SMO | Leukemia | Phase 2 | NCT01357655 | Dasatinib | US | Terminated |
| Phase 1, 2 | NCT01218477 | Dasatinib | US | Completed |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sari, I.N.; Phi, L.T.H.; Jun, N.; Wijaya, Y.T.; Lee, S.; Kwon, H.Y. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells 2018, 7, 208. https://doi.org/10.3390/cells7110208
Sari IN, Phi LTH, Jun N, Wijaya YT, Lee S, Kwon HY. Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells. 2018; 7(11):208. https://doi.org/10.3390/cells7110208
Chicago/Turabian StyleSari, Ita Novita, Lan Thi Hanh Phi, Nayoung Jun, Yoseph Toni Wijaya, Sanghyun Lee, and Hyog Young Kwon. 2018. "Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells" Cells 7, no. 11: 208. https://doi.org/10.3390/cells7110208
APA StyleSari, I. N., Phi, L. T. H., Jun, N., Wijaya, Y. T., Lee, S., & Kwon, H. Y. (2018). Hedgehog Signaling in Cancer: A Prospective Therapeutic Target for Eradicating Cancer Stem Cells. Cells, 7(11), 208. https://doi.org/10.3390/cells7110208