Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy


Abstract
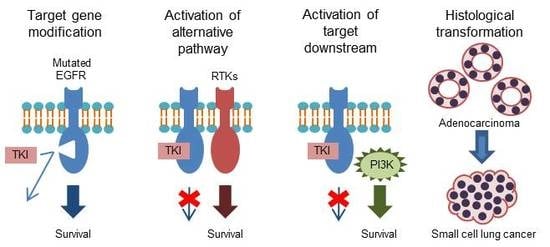
:
{kind=link}
{kind=link}
{kind=link}
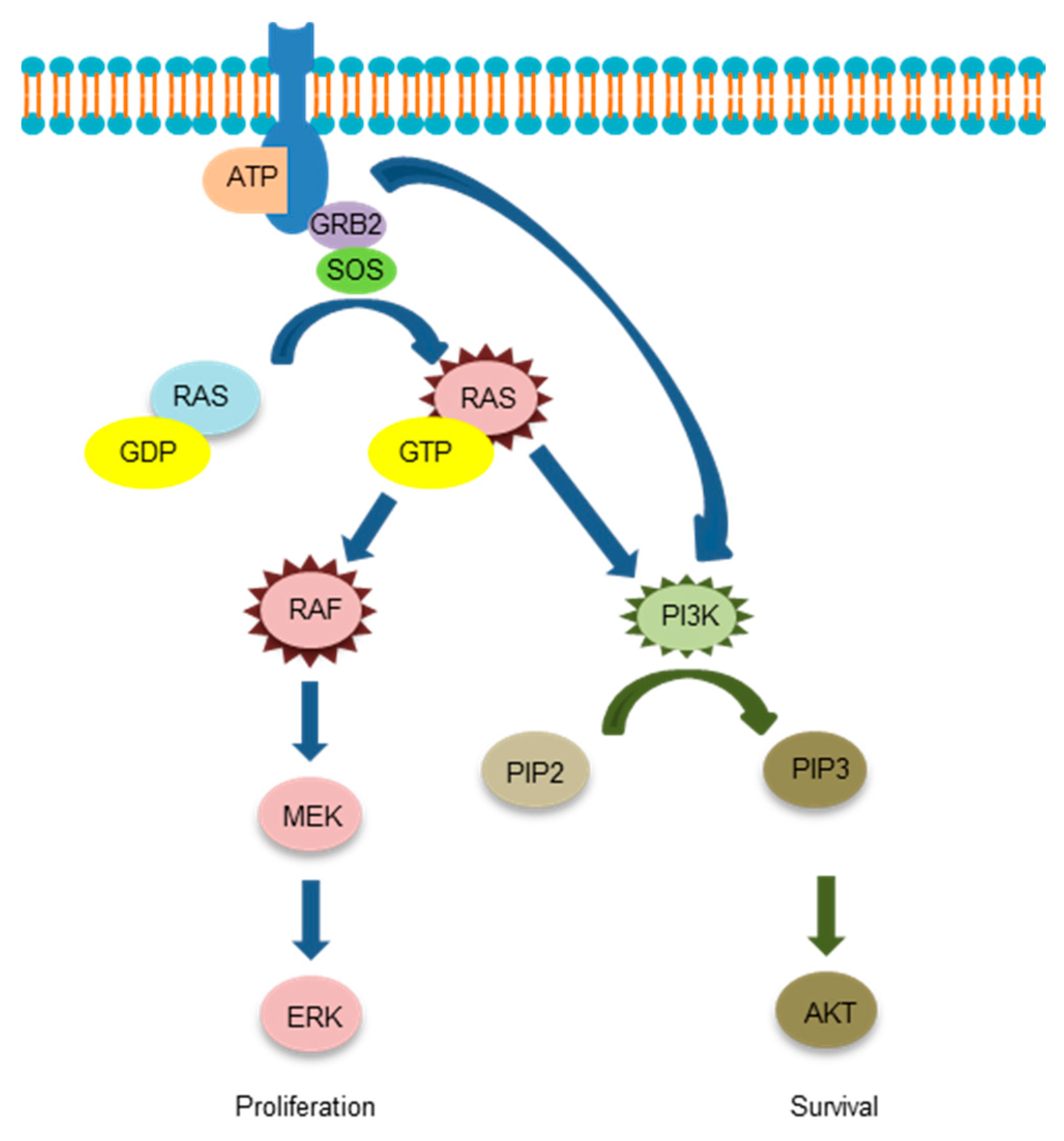{kind=link}
1. Introduction
2. Acquired Resistance
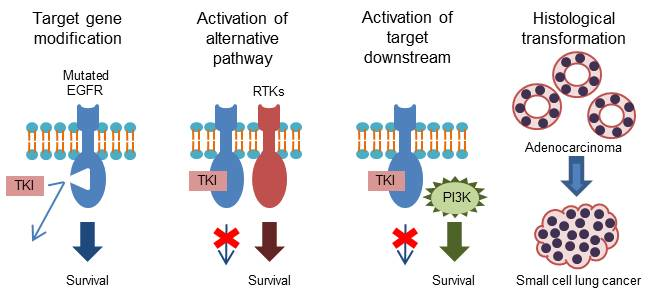
2.1. T790M “Gatekeeper” Mutation
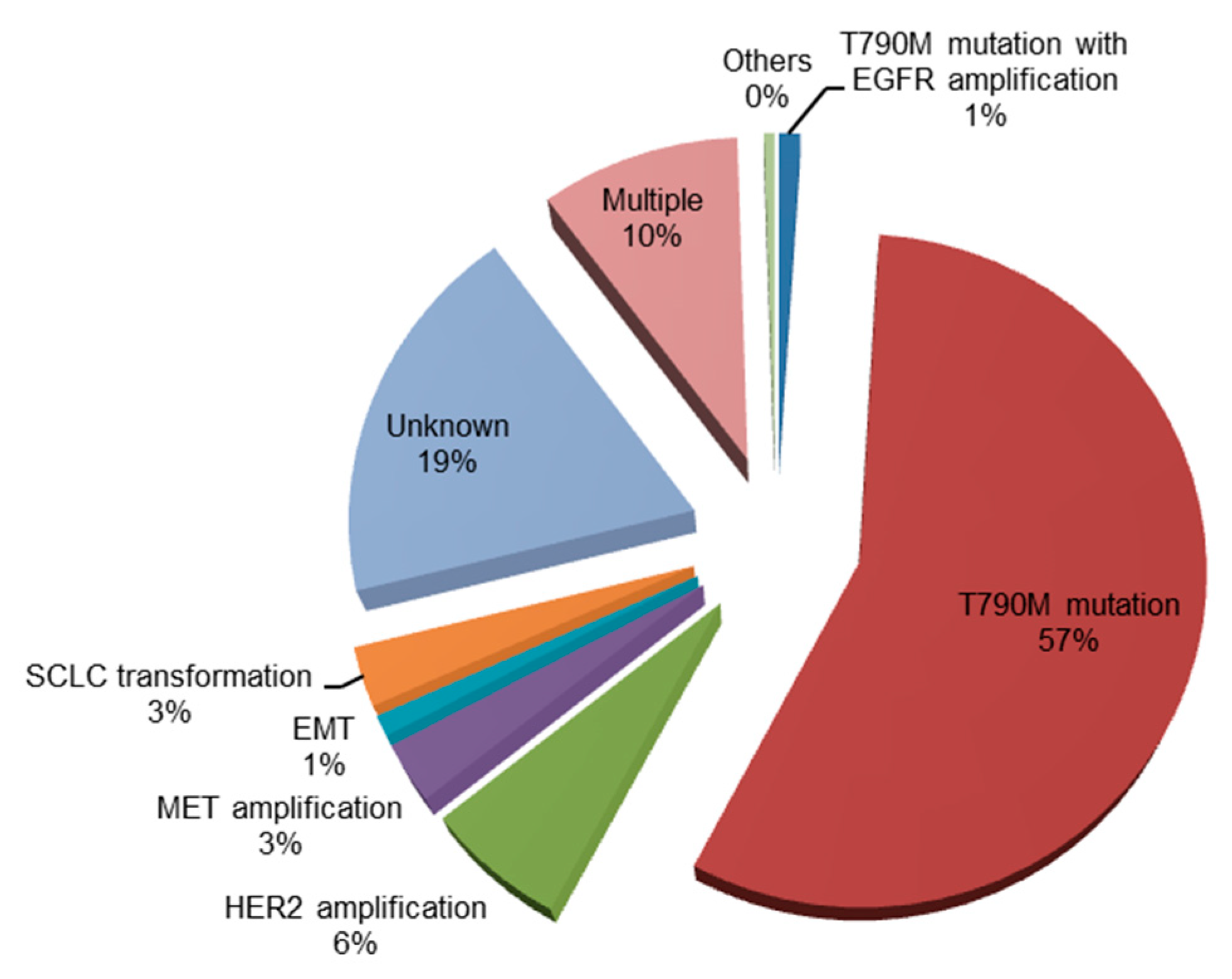
2.2. Acquired Resistance to Osimertinib
2.2.1. C797S Mutation
2.2.2. Other EGFR Mutation
2.2.3. Oncogenic Fusion
2.2.4. Cell Cycle Gene Alterations
2.2.5. MAPK/PI3K alterations
2.2.6. Others
2.3. Activation of Alternative Pathways
2.3.1. MET Gene Amplification
2.3.2. HGF Overexpression
2.3.3. Insulin-Like Growth Factor (IGF) Upregulation
2.3.4. HER2 Amplification
2.3.5. Growth Arrest-Specific 6 (GAS6)/ Anexelekto (AXL) Activation
2.4. Activation of Downstream Targets
2.4.1. KRAS Mutation
2.4.2. BRAF Mutation
2.4.3. PIK3CA Mutation
2.4.4. Phosphatase and Tensin Homolog (PTEN) Deletion
2.4.5. NF-1 Deletion
2.4.6. v-crk Sarcoma Virus CT10 Oncogene Homolog (Avian)-Like (CRKL) Amplification
2.5. Histologic Transformation
2.5.1. EMT
2.5.2. SCLC Transformation
3. Primary Resistance
3.1. HGF Overexpression
3.2. Decrease of BIM
3.3. Others
4. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Togashi, Y.; Yatabe, Y.; Mizuuchi, H.; Jangchul, P.; Kondo, C.; Shimoji, M.; Sato, K.; Suda, K.; Tomizawa, K.; et al. EGFR exon 18 mutations in lung cancer: Molecular predictors of augmented sensitivity to afatinib or neratinib as compared with first- or third-generation TKIs. Clin. Cancer Res. 2015, 21, 5305–5313. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tam, K.Y. Combination strategies using EGFR-TKi in NSCLC therapy: Learning from the gap between pre-clinical results and clinical outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Campo, M.; Gerber, D.; Gainor, J.F.; Heist, R.S.; Temel, J.S.; Shaw, A.T.; Fidias, P.; Muzikansky, A.; Engelman, J.A.; Sequist, L.V. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J. Thorac. Oncol. 2016, 11, 2022–2026. [Google Scholar] [CrossRef] [PubMed]
- Proto, C.; Lo Russo, G.; Corrao, G.; Ganzinelli, M.; Facchinetti, F.; Minari, R.; Tiseo, M.; Garassino, M.C. Treatment in EGFR-mutated non-small cell lung cancer: How to block the receptor and overcome resistance mechanisms. Tumori 2017, 103, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Moran, T.; Queralt, C.; Porta, R.; Cardenal, F.; Camps, C.; Majem, M.; Lopez-Vivanco, G.; Isla, D.; Provencio, M.; et al. Screening for epidermal growth factor receptor mutations in lung cancer. N. Engl. J. Med. 2009, 361, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Martins, R.G.; Spigel, D.; Grunberg, S.M.; Spira, A.; Jänne, P.A.; Joshi, V.A.; McCollum, D.; Evans, T.L.; Muzikansky, A.; et al. First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J. Clin. Oncol. 2008, 26, 2442–2449. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Wang, J.; Zhang, S. Mechanisms of resistance to irreversible epidermal growth factor receptor tyrosine kinase inhibitors and therapeutic strategies in non-small cell lung cancer. Oncotarget 2017, 8, 90557–90578. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Yang, J.C.; Yamamoto, N.; O′Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.M.; Boyer, M.; et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhou, C.; Hu, C.P.; Feng, J.; Lu, S.; Huang, Y.; Li, W.; Hou, M.; Shi, J.H.; Lee, K.Y.; et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): An open-label, randomised phase 3 trial. Lancet Oncol. 2014, 15, 213–222. [Google Scholar] [CrossRef]
- Park, K.; Tan, E.H.; O’Byrne, K.; Zhang, L.; Boyer, M.; Mok, T.; Hirsh, V.; Yang, J.C.; Lee, K.H.; Lu, S.; et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016, 17, 577–589. [Google Scholar] [CrossRef]
- Wu, S.G.; Liu, Y.N.; Tsai, M.F.; Chang, Y.L.; Yu, C.J.; Yang, P.C.; Yang, J.C.; Wen, Y.F.; Shih, J.Y. The mechanism of acquired resistance to irreversible EGFR tyrosine kinase inhibitor-afatinib in lung adenocarcinoma patients. Oncotarget 2016, 7, 12404–12413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katakami, N.; Atagi, S.; Goto, K.; Hida, T.; Horai, T.; Inoue, A.; Ichinose, Y.; Koboyashi, K.; Takeda, K.; Kiura, K.; et al. LUX-Lung 4: A phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J. Clin. Oncol. 2013, 31, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [PubMed]
- Soejima, K.; Yasuda, H.; Hirano, T. Osimertinib for EGFR T790M mutation-positive non-small cell lung cancer. Expert Rev. Clin. Pharmacol. 2017, 10, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Yasuda, H.; Tani, T.; Hamamoto, J.; Oashi, A.; Ishioka, K.; Arai, D.; Nukaga, S.; Miyawaki, M.; Kawada, I.; et al. In vitro modeling to determine mutation specificity of EGFR tyrosine kinase inhibitors against clinically relevant EGFR mutants in non-small-cell lung cancer. Oncotarget 2015, 6, 38789–38803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jänne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.L.; Ahn, M.J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or platinum-pemetrexed in EGFR T790M-positive lung cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of acquired resistance to AZD9291: A mutation-selective, irreversible EGFR inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Thress, K.S.; Mooradian, M.; Heist, R.S.; Azzoli, C.G.; Temel, J.S.; Rizzo, C.; Nagy, R.J.; Lanman, R.B.; Gettinger, S.N.; et al. MET amplification (amp) as a resistance mechanism to osimertinib. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Guibert, N.M.; Paweletz, C.; Hu, Y.; Feeney, N.B.; Plagnol, V.; Poole, V.; Jones, G.; Oxnard, G.R. Early detection of competing resistance mutations using plasma next-generation sequencing (NGS) in patients (pts) with EGFR-mutant NSCLC treated with osimertinib. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Ou, Q.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Zhang, X.; Chang, Z.; Mao, Y.; Shao, Y.; Zhou, C. Investigating novel resistance mechanisms to third generation EGFR TKI osimertinib in non-small cell lung cancer patients using next generation sequencing. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Costa, D.B.; Kobayashi, S.S. Whacking a mole-cule: Clinical activity and mechanisms of resistance to third generation EGFR inhibitors in EGFR mutated lung cancers with EGFR-T790M. Transl. Lung Cancer Res. 2015, 4, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.A.; Tian, S.K.; Drilon, A.E.; Borsu, L.; Riely, G.J.; Arcila, M.E.; Ladanyi, M. Acquired Resistance of EGFR-Mutant Lung Cancer to a T790M-Specific EGFR Inhibitor: Emergence of a Third Mutation (C797S) in the EGFR Tyrosine Kinase Domain. JAMA Oncol. 2015, 1, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Cuaran, S.; Scheffler, M.; Plenker, D.; Dahmen, L.; Scheel, A.H.; Fernandez-Cuesta, L.; Meder, L.; Lovly, C.M.; Persigehl, T.; Merkelbach-Bruse, S.; et al. Heterogeneous mechanisms of primary and acquired resistance to third-generation EGFR inhibitors. Clin. Cancer Res. 2016, 22, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Ercan, D.; Choi, H.G.; Yun, C.H.; Capelletti, M.; Xie, T.; Eck, M.J.; Gray, N.S.; Jänne, P.A. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Simmons, A.D.; Lovejoy, A.F.; Esfahani, M.S.; Newman, A.M.; Haringsma, H.J.; Kurtz, D.M.; Stehr, H.; Scherer, F.; Karlovich, C.A.; et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat. Commun. 2016, 7, 11815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.N.; Jung, K.S.; Yoo, K.H.; Cho, J.; Lee, J.Y.; Lim, S.H.; Kim, H.S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Acquired C797S mutation upon treatment with a T790M-specific third-generation EGFR inhibitor (HM61713) in non-small cell lung cancer. J. Thorac. Oncol. 2016, 11, e45–e47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, J.J.; Huang, J.; Ye, J.Y.; Zhang, X.C.; Tu, H.Y.; Han-Zhang, H.; Wu, Y.L. Lung adenocarcinoma harboring EGFR T790M and in trans C797S responds to combination therapy of first- and third-generation EGFR TKIs and shifts allelic configuration at resistance. J. Thorac. Oncol. 2017, 12, 1723–1727. [Google Scholar] [CrossRef] [PubMed]
- Arulananda, S.; Do, H.; Musafer, A.; Mitchell, P.; Dobrovic, A.; John, T. Combination osimertinib and gefitinib in C797S and T790M EGFR-mutated non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 1728–1732. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Chen, L.; Sangji, N.; Okabe, T.; Yonesaka, K.; Francis, J.M.; Flavin, R.J.; Johnson, W.; Kwon, J.; Yu, S.; et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013, 73, 6770–6779. [Google Scholar] [CrossRef] [PubMed]
- Uchibori, K.; Inase, N.; Araki, M.; Kamada, M.; Sato, S.; Okuno, Y.; Fujita, N.; Katayama, R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, Y.; Yun, C.H.; Park, E.; Ercan, D.; Manuia, M.; Juarez, J.; Xu, C.; Rhee, K.; Chen, T.; Zhang, H.; et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534, 129–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bersanelli, M.; Minari, R.; Bordi, P.; Gnetti, L.; Bozzetti, C.; Squadrilli, A.; Lagrasta, C.A.; Bottarelli, L.; Osipova, G.; Capelletto, E.; et al. L718Q mutation as new mechanism of acquired resistance to AZD9291 in EGFR-mutated NSCLC. J. Thorac. Oncol. 2016, 11, e121–e123. [Google Scholar] [CrossRef] [PubMed]
- Papadimitrakopoulou, V.A.; Wu, Y.; Han, J.; Ahn, M.; Ramalingam, S.S.; John, T.; Okamoto, I.; Yang, J.C.; Bulusu, K.C.; Laus, G.; et al. Analysis of resistance mechanisms to osimertinib in patients with EGFR T790M advanced NSCLC from the AURA3 study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Eck, M.J.; Yun, C.H. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim. Biophys. Acta 2010, 1804, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29. [Google Scholar] [CrossRef]
- Ying, J.; Lin, C.; Wu, J.; Guo, L.; Qiu, T.; Ling, Y.; Shan, L.; Zhou, H.; Zhao, D.; Wang, J.; et al. Anaplastic lymphoma kinase rearrangement in digestive tract cancer: Implication for targeted therapy in chinese population. PLoS ONE 2015, 10, e0144731. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.F.; Zhang, Y.; Liu, Y.Y.; Hong, X.H.; Liang, J.Y.; Tong, F.; Yang, J.S.; Liu, L. Lung adenocarcinoma harboring concomitant SPTBN1-ALK fusion, c-Met overexpression, and HER-2 amplification with inherent resistance to crizotinib, chemotherapy, and radiotherapy. J. Hematol. Oncol. 2016, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.C.; Liao, W.Y.; Lin, C.A.; Shih, J.Y.; Yu, C.J.; Chih-Hsin Yang, J. Acquired BRAF V600E mutation as resistant mechanism after treatment with osimertinib. J. Thorac. Oncol. 2017, 12, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Planchard, D.; Loriot, Y.; André, F.; Gobert, A.; Auger, N.; Lacroix, L.; Soria, J.C. EGFR-independent mechanisms of acquired resistance to AZD9291 in EGFR T790M-positive NSCLC patients. Ann. Oncol. 2015, 26, 2073–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukaga, S.; Yasuda, H.; Tsuchihara, K.; Hamamoto, J.; Masuzawa, K.; Kawada, I.; Naoki, K.; Matsumoto, S.; Mimaki, S.; Ikemura, S.; et al. Amplification of EGFR wild-type alleles in non-small cell lung cancer cells confers acquired resistance to mutation-selective EGFR tyrosine kinase inhibitors. Cancer Res. 2017, 77, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Bean, J.; Brennan, C.; Shih, J.Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Oh, Y.T.; Zhang, G.; Yao, W.; Yue, P.; Li, Y.; Kanteti, R.; Riehm, J.; Salgia, R.; Owonikoko, T.K.; et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016, 380, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Niederst, M.J.; Lennerz, J.K.; Dagogo-Jack, I.; Stevens, S.; Shaw, A.T.; Sequist, L.V.; Engelman, J.A. Dramatic response to combination erlotinib and crizotinib in a patient with advanced, EGFR-mutant lung cancer harboring de novo MET amplification. J. Thorac. Oncol. 2016, 11, e83–e85. [Google Scholar] [CrossRef] [PubMed]
- York, E.R.; Varella-Garcia, M.; Bang, T.J.; Aisner, D.L.; Camidge, D.R. Tolerable and effective combination of full-dose crizotinib and osimertinib targeting MET amplification sequentially emerging after T790M positivity in EGFR-mutant non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, e85–e88. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Zhang, L.; Kim, D.W.; Liu, X.; Lee, D.H.; Yang, J.C.; Ahn, M.J.; Vansteenkiste, J.F.; Su, W.C.; Felip, E.; et al. Phase Ib/II study of capmatinib (INC280) plus gefitinib after failure of epidermal growth factor receptor (EGFR) inhibitor therapy in patients with EGFR-mutated, MET factor-dysregulated non-small-cell lung cancer. J. Clin. Oncol. 2018, JCO2018777326. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Kim, J.Y.; Lee, S.H.; Yoo, N.J.; Choi, B.G. Association between plasma hepatocyte growth factor and gefitinib resistance in patients with advanced non-small cell lung cancer. Lung Cancer 2011, 74, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Masago, K.; Togashi, Y.; Fujita, S.; Sakamori, Y.; Okuda, C.; Kim, Y.H.; Mio, T.; Mishima, M. Clinical significance of serum hepatocyte growth factor and epidermal growth factor gene somatic mutations in patients with non-squamous non-small cell lung cancer receiving gefitinib or erlotinib. Med. Oncol. 2012, 29, 1614–1621. [Google Scholar] [CrossRef] [PubMed]
- Arrieta, O.; Cruz-Rico, G.; Soto-Perez-de-Celis, E.; Ramírez-Tirado, L.A.; Caballe-Perez, E.; Martínez-Hernández, J.N.; Martinez-Alvarez, I.; Soca-Chafre, G.; Macedo-Pérez, E.O.; Astudillo-de la Vega, H. Reduction in hepatocyte growth factor serum levels is associated with improved prognosis in advanced lung adenocarcinoma patients treated with afatinib: A phase II trial. Target. Oncol. 2016, 11, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Wang, W.; Li, Q.; Matsumoto, K.; Sakurama, H.; Nakamura, T.; Ogino, H.; Kakiuchi, S.; Hanibuchi, M.; Nishioka, Y.; et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008, 68, 9479–9487. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Hashimoto, E.; Nakatani, N.; Abe, M.; Satoh, Y.; Sakata, K.; Fujii, T.; Fujimoto-Ouchi, K.; Sugimoto, M.; Nagahashi, S.; et al. Combining onartuzumab with erlotinib inhibits growth of non-small cell lung cancer with activating EGFR mutations and HGF overexpression. Mol. Cancer Ther. 2015, 14, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Cappuzzo, F.; Tallini, G.; Finocchiaro, G.; Wilson, R.S.; Ligorio, C.; Giordano, L.; Toschi, L.; Incarbone, M.; Cavina, R.; Terracciano, L.; et al. Insulin-like growth factor receptor 1 (IGF1R) expression and survival in surgically resected non-small-cell lung cancer (NSCLC) patients. Ann. Oncol. 2010, 21, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Wang, Y.; James, M.; Jeong, J.H.; You, M. Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells. Mol. Carcinog. 2016, 55, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Choi, Y.J.; Kim, S.Y.; Lee, J.E.; Sung, K.J.; Park, S.; Kim, W.S.; Song, J.S.; Choi, C.M.; Sung, Y.H.; et al. Activation of the IGF1R pathway potentially mediates acquired resistance to mutant-selective 3rd-generation EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 22005–22015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.P.; Hastings, J.F.; Han, J.Z.; Croucher, D.R. The under-appreciated promiscuity of the epidermal growth factor receptor family. Front. Cell Dev. Biol. 2016, 4, 88. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef] [PubMed]
- Takezawa, K.; Pirazzoli, V.; Arcila, M.E.; Nebhan, C.A.; Song, X.; de Stanchina, E.; Ohashi, K.; Janjigian, Y.Y.; Spitzler, P.J.; Melnick, M.A.; et al. HER2 amplification: A potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer Discov. 2012, 2, 922–933. [Google Scholar] [CrossRef] [PubMed]
- Pirazzoli, V.; Ayeni, D.; Meador, C.B.; Sanganahalli, B.G.; Hyder, F.; de Stanchina, E.; Goldberg, S.B.; Pao, W.; Politi, K. Afatinib plus cetuximab delays resistance compared to single-agent erlotinib or afatinib in mouse models of TKI-naïve EGFR L858R-induced lung adenocarcinoma. Clin. Cancer Res. 2016, 22, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Gatzemeier, U.; Groth, G.; Butts, C.; Van Zandwijk, N.; Shepherd, F.; Ardizzoni, A.; Barton, C.; Ghahramani, P.; Hirsh, V. Randomized phase II trial of gemcitabine-cisplatin with or without trastuzumab in HER2-positive non-small-cell lung cancer. Ann. Oncol. 2004, 15, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazières, J.; Barlesi, F.; Filleron, T.; Besse, B.; Monnet, I.; Beau-Faller, M.; Peters, S.; Dansin, E.; Früh, M.; Pless, M.; et al. Lung cancer patients with HER2 mutations treated with chemotherapy and HER2-targeted drugs: Results from the European EUHER2 cohort. Ann. Oncol. 2016, 27, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Langen, J.D.; Kuiper, J.L.; Thunnissen, E.; Hashemi, S.M.; Monkhorst, K.; Smit, E.F. Trastuzumab and paclitaxel in patients (pts) with EGFR mutated non-small-cell lung cancer (NSCLC) that express HER2 after progression on EGFR TKI treatment. J. Clin. Oncol. 2017, 35. [Google Scholar] [CrossRef]
- Zhang, Z.; Lee, J.C.; Lin, L.; Olivas, V.; Au, V.; LaFramboise, T.; Abdel-Rahman, M.; Wang, X.; Levine, A.D.; Rho, J.K.; et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat. Genet. 2012, 44, 852–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, C.M.; Balaji, K.; Byers, L.A. Giving AXL the axe: Targeting AXL in human malignancy. Br. J. Cancer 2017, 116, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.M.; Kim, H.R.; Cho, E.K.; Min, Y.J.; Ahn, J.S.; Ahn, M.J.; Park, K.; Cho, B.C.; Lee, J.H.; Jeong, H.C.; et al. Targeted sequencing identifies genetic alterations that confer primary resistance to EGFR tyrosine kinase inhibitor (Korean Lung Cancer Consortium). Oncotarget 2016, 7, 36311–36320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren-Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, T.; Kang, P.; Qian, K.; Deng, B.; Zhou, J.; Wang, R.; Jiang, B.; Li, K.; Liu, F.; et al. Combined analysis of rearrangement of ALK, ROS1, somatic mutation of EGFR, KRAS, BRAF, PIK3CA, and mRNA expression of ERCC1, TYMS, RRM1, TUBB3, EGFR in patients with non-small cell lung cancer and their clinical significance. Cancer Chemother. Pharmacol. 2016, 77, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yin, W.; He, W.; Jiang, C.; Zhou, X.; Song, X.; Zhu, J.; Fei, K.; Cao, W.; Jiang, G. Phenotype-genotype correlation in multiple primary lung cancer patients in China. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Fan, P.D.; Shen, R.; Zakowski, M.; Varmus, H. Erlotinib resistance in mouse models of epidermal growth factor receptor-induced lung adenocarcinoma. Dis. Model. Mech. 2010, 3, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Moran, T.; Chmielecki, J.; Lin, Y.L.; Pan, Y.; Wang, L.; de Stanchina, E.; Shien, K.; et al. Lung cancers with acquired resistance to EGFR inhibitors occasionally harbor BRAF gene mutations but lack mutations in KRAS, NRAS, or MEK1. Proc. Natl. Acad. Sci. USA 2012, 109, E2127–E2133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, Y.; Takano, O.; Kato, I.; Takahashi, Y.; Shima, F.; Kataoka, T. Ras inhibitors display an anti-metastatic effect by downregulation of lysyl oxidase through inhibition of the Ras-PI3K-Akt-HIF-1α pathway. Cancer Lett. 2017, 410, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, Q.; Zhang, S.; Chen, J.; Huang, L.; Ren, J.; Chang, Y.; Liang, Y.; Wu, G. NVP-BEZ235 overcomes gefitinib-acquired resistance by down-regulating PI3K/AKT/mTOR phosphorylation. Onco Targets Ther. 2015, 8, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Lammers, P.E.; Lovly, C.M.; Horn, L. A patient with metastatic lung adenocarcinoma harboring concurrent EGFR L858R, EGFR germline T790M, and PIK3CA mutations: The challenge of interpreting results of comprehensive mutational testing in lung cancer. J. Natl. Compr. Cancer Netw. 2014, 12, 6–11. [Google Scholar] [CrossRef]
- Lara, P.N.; Longmate, J.; Mack, P.C.; Kelly, K.; Socinski, M.A.; Salgia, R.; Gitlitz, B.; Li, T.; Koczywas, M.; Reckamp, K.L.; et al. Phase II study of the AKT inhibitor MK-2206 plus erlotinib in patients with advanced non-small cell lung cancer who previously progressed on erlotinib. Clin. Cancer Res. 2015, 21, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.C.; Lee, H.Y.; Lee, J.I.; Wang, L.; Issa, J.P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Stambolic, V.; Suzuki, A.; de la Pompa, J.L.; Brothers, G.M.; Mirtsos, C.; Sasaki, T.; Ruland, J.; Penninger, J.M.; Siderovski, D.P.; Mak, T.W. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 1998, 95, 29–39. [Google Scholar] [CrossRef]
- Yamasaki, F.; Johansen, M.J.; Zhang, D.; Krishnamurthy, S.; Felix, E.; Bartholomeusz, C.; Aguilar, R.J.; Kurisu, K.; Mills, G.B.; Hortobagyi, G.N.; et al. Acquired resistance to erlotinib in A-431 epidermoid cancer cells requires down-regulation of MMAC1/PTEN and up-regulation of phosphorylated Akt. Cancer Res. 2007, 67, 5779–5788. [Google Scholar] [CrossRef] [PubMed]
- She, Q.B.; Solit, D.B.; Ye, Q.; O’Reilly, K.E.; Lobo, J.; Rosen, N. The BAD protein integrates survival signaling by EGFR/MAPK and PI3K/Akt kinase pathways in PTEN-deficient tumor cells. Cancer Cell 2005, 8, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sos, M.L.; Koker, M.; Weir, B.A.; Heynck, S.; Rabinovsky, R.; Zander, T.; Seeger, J.M.; Weiss, J.; Fischer, F.; Frommolt, P.; et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009, 69, 3256–3261. [Google Scholar] [CrossRef] [PubMed]
- To, K.K.W.; Wu, W.K.K.; Loong, H.H.F. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur. J. Pharmacol. 2018, 823, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Le, L.Q.; Parada, L.F. Tumor microenvironment and neurofibromatosis type I: Connecting the GAPs. Oncogene 2007, 26, 4609–4616. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Yuan, C.; Cheng, C.; Zhang, Y.; Ma, Y.; Zheng, D.; Zheng, S.; Li, Y.; Jin, Y.; Sun, Y.; et al. Frequency and clinical significance of NF1 mutation in lung adenocarcinomas from East Asian patients. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheung, H.W.; Du, J.; Boehm, J.S.; He, F.; Weir, B.A.; Wang, X.; Butaney, M.; Sequist, L.V.; Luo, B.; Engelman, J.A.; et al. Amplification of CRKL induces transformation and epidermal growth factor receptor inhibitor resistance in human non-small cell lung cancers. Cancer Discov. 2011, 1, 608–625. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Frederick, B.A.; Helfrich, B.A.; Coldren, C.D.; Zheng, D.; Chan, D.; Bunn, P.A.; Raben, D. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol. Cancer Ther. 2007, 6, 1683–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rho, J.K.; Choi, Y.J.; Lee, J.K.; Ryoo, B.Y.; Na, I.I.; Yang, S.H.; Kim, C.H.; Lee, J.C. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer 2009, 63, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.H.; Rho, J.K.; Xu, X.; Lee, J.S.; Yoon, H.I.; Lee, C.T.; Choi, Y.J.; Kim, H.R.; Kim, C.H.; Lee, J.C. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011, 73, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Epstein, D.; Haley, J.D. Kinase switching in mesenchymal-like non-small cell lung cancer lines contributes to EGFR inhibitor resistance through pathway redundancy. Clin. Exp. Metast. 2008, 25, 843–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Murakami, A.; Takahashi, K. Epithelial mesenchymal transition in drug resistance and metastasis of lung cancer. Cancer Res. Treat. 2012, 44, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Lee, J.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; Kim, Y.T.; et al. Clonal history and genetic predictors of transformation into small-cell Carcinomas from lung adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.S.; Kim, S.; Kim, H.K.; Byeon, S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Park, K.; Choi, Y.L.; Han, J.; et al. Two cases of small cell lung cancer transformation from EGFR mutant adenocarcinoma during AZD9291 treatment. J. Thorac. Oncol. 2016, 11, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Matsumoto, K.; Wang, W.; Li, Q.; Nishioka, Y.; Sekido, Y.; Sone, S.; Yano, S. Hepatocyte growth factor reduces susceptibility to an irreversible epidermal growth factor receptor inhibitor in EGFR-T790M mutant lung cancer. Clin. Cancer Res. 2010, 16, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Faber, A.C.; Corcoran, R.B.; Ebi, H.; Sequist, L.V.; Waltman, B.A.; Chung, E.; Incio, J.; Digumarthy, S.R.; Pollack, S.F.; Song, Y.; et al. BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011, 1, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Molina, M.A.; Drozdowskyj, A.; Giménez-Capitán, A.; Bertran-Alamillo, J.; Karachaliou, N.; Gervais, R.; Massuti, B.; Wei, J.; Moran, T.; et al. The impact of EGFR T790M mutations and BIM mRNA expression on outcome in patients with EGFR-mutant NSCLC treated with erlotinib or chemotherapy in the randomized phase III EURTAC trial. Clin. Cancer Res. 2014, 20, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Greulich, H.; Chen, T.H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R.; et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005, 2, 1167–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hu, H.; Pan, Y.; Wang, R.; Li, Y.; Shen, L.; Yu, Y.; Li, H.; Cai, D.; Sun, Y.; et al. PIK3CA mutations frequently coexist with EGFR/KRAS mutations in non-small cell lung cancer and suggest poor prognosis in EGFR/KRAS wildtype subgroup. PLoS ONE 2014, 9, e88291. [Google Scholar] [CrossRef] [PubMed]
- Sos, M.L.; Zander, T.; Thomas, R.K.; Staratschek-Jox, A.; Claasen, J.; Wolf, J. Expression of signaling mediators downstream of EGF-receptor predict sensitivity to small molecule inhibitors directed against the EGF-receptor pathway. J. Thorac. Oncol. 2008, 3, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.G.; Arcila, M.E.; Yu, H.A. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. https://doi.org/10.3390/cells7110212
Nagano T, Tachihara M, Nishimura Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells. 2018; 7(11):212. https://doi.org/10.3390/cells7110212
Chicago/Turabian StyleNagano, Tatsuya, Motoko Tachihara, and Yoshihiro Nishimura. 2018. "Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy" Cells 7, no. 11: 212. https://doi.org/10.3390/cells7110212
APA StyleNagano, T., Tachihara, M., & Nishimura, Y. (2018). Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells, 7(11), 212. https://doi.org/10.3390/cells7110212