Proteomic Signatures of Clostridium difficile Stressed with Metronidazole, Vancomycin, or Fidaxomicin

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
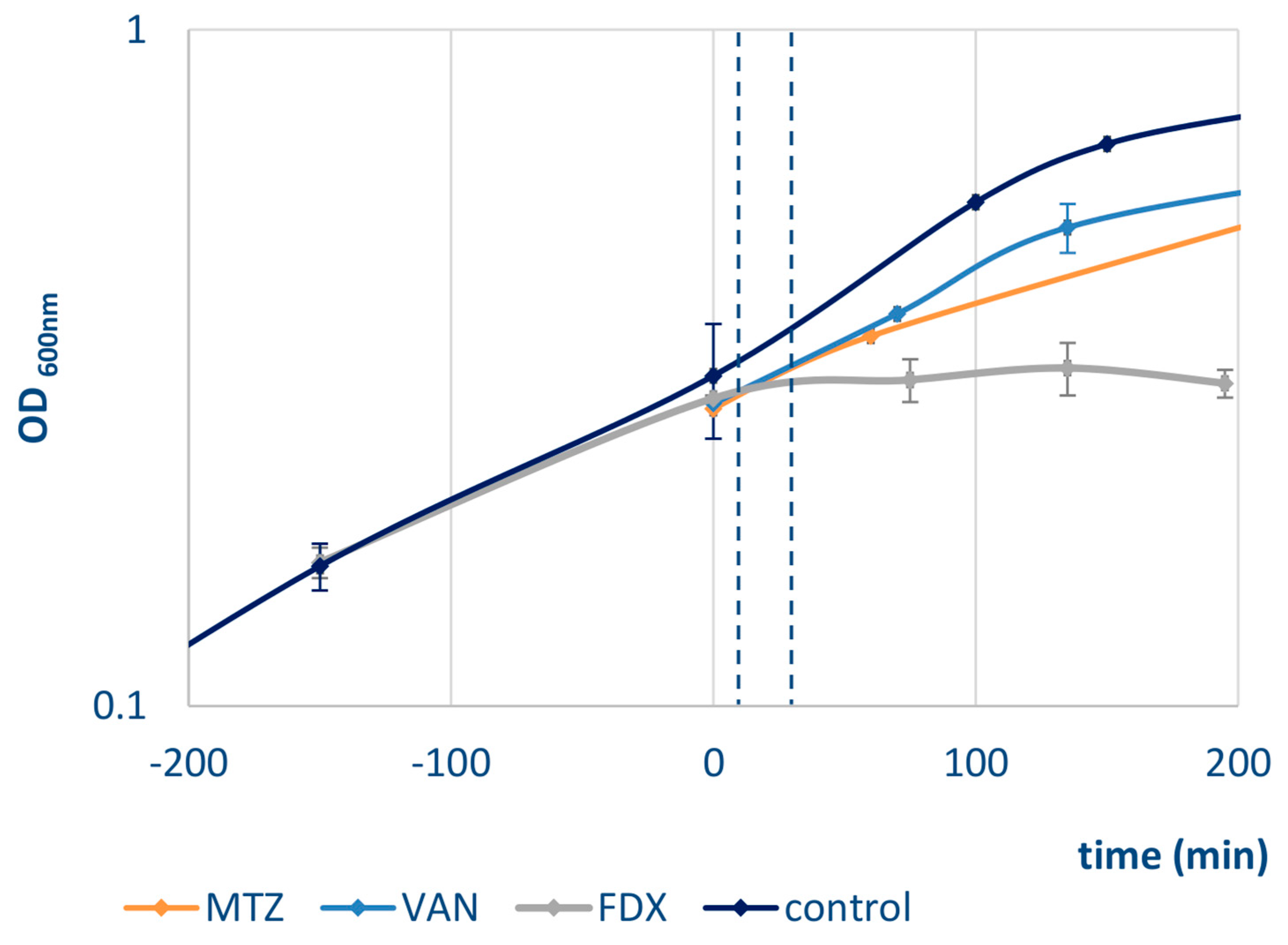
2.1. Bacterial Growth and Antibiotic Stress
2.2. Radioactive Pulse-Labeling, Cell Harvest, and Protein Preparation
2.3. 2D PAGE and Autoradiography
2.4. Processing of Gel Images and Statistical Analysis
2.5. Spot Identification Using MALDI-TOF-MS
3. Results
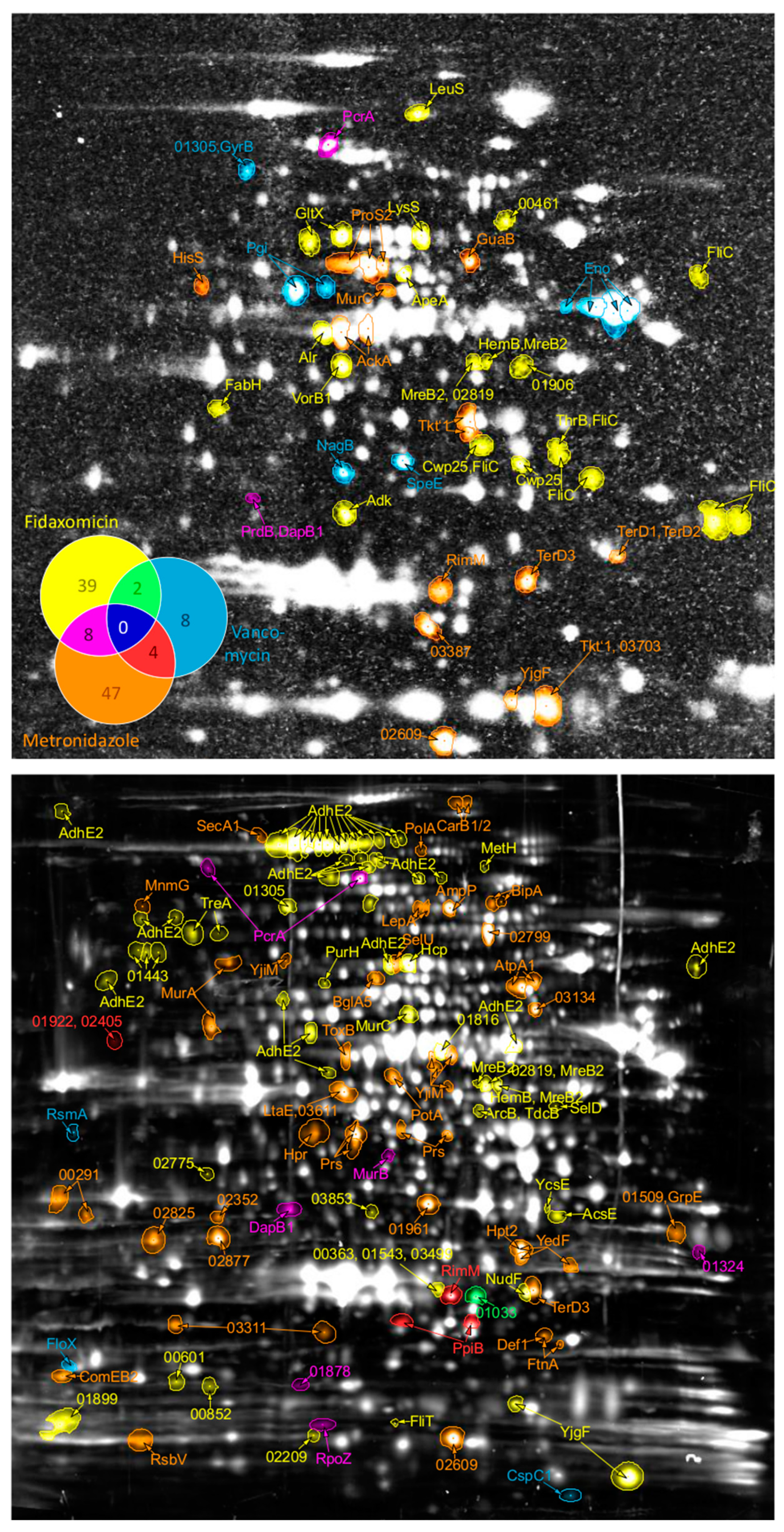
3.1. Proteomic Signatures for Vancomycin, Metronidazole, and Fidaxomicin Stress
3.2. Proteomic Adaptation to Vancomycin
3.3. Proteomic Adaptation to Metronidazole
3.4. Proteomic Adaptation to Fidaxomicin
4. Discussion
4.1. Technical Aspects of Gel-Based Approaches to Derive Antibiotic Signature
4.2. Protein Signatures in Antibiotic Research in C. difficile
4.3. Response to Metronidazole in Other Anaerobic Bacteria
4.4. Detoxification of Antimicrobial Compounds in C. difficile
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lawson, P.A.; Citron, D.M.; Tyrrell, K.L.; Finegold, S.M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 2016, 40, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Mergenhagen, K.A.; Wojciechowski, A.L.; Paladino, J.A. A review of the economics of treating Clostridium difficile infection. Pharmacoeconomics 2014, 32, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.H.; Howe, R. Diarrhoea caused by Clostridium difficile: Response time for treatment with metronidazole and vancomycin. J. Antimicrob. Chemother. 1995, 36, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N. Treatment of Clostridium difficile-associated diarrhea and colitis. Curr. Top. Microbiol. Immunol. 2000, 250, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Surawicz, C.M.; Brandt, L.J.; Binion, D.G.; Ananthakrishnan, A.N.; Curry, S.R.; Gilligan, P.H.; McFarland, L.V.; Mellow, M.; Zuckerbraun, B.S. Guidelines for diagnosis, treatment, and prevention of Clostridium difficile infections. Am. J. Gastroenterol. 2013, 108, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Perkins, H.R. Specificity of combination between mucopeptide precursors and vancomycin or ristocetin. Biochem. J. 1969, 111, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerding, D.N. Is there a relationship between vancomycin-resistant enterococcal infection and Clostridium difficile infection? Clin. Infect. Dis. 1997, 25, S206–S210. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M.; Spengler, G.; Urbán, E. Identification and antimicrobial susceptibility testing of anaerobic bacteria: Rubik’s cube of clinical microbiology? Antibiotics 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R.; Murray, B.E.; Rice, L.B.; Arias, C.A. Vancomycin-resistant enterococci: Therapeutic challenges in the 21st century. Infect. Dis. Clin. N. Am. 2016, 30, 415–439. [Google Scholar] [CrossRef] [PubMed]
- Barbut, F.; Decré, D.; Burghoffer, B.; Lesage, D.; Delisle, F.; Lalande, V.; Delmée, M.; Avesani, V.; Sano, N.; Coudert, C.; et al. Antimicrobial susceptibilities and serogroups of clinical strains of Clostridium difficile isolated in France in 1991 and 1997. Antimicrob. Agents Chemother. 1999, 43, 2607–2611. [Google Scholar] [CrossRef] [PubMed]
- Peláez, T.; Alcalá, L.; Alonso, R.; Rodríguez-Créixems, M.; García-Lechuz, J.M.; Bouza, E. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob. Agents Chemother. 2002, 46, 1647–1650. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.-Y.; Woo, P.C.-Y.; Luk, W.-K.; Yuen, K.-Y. Susceptibility testing of Clostridium difficile against metronidazole and vancomycin by disk diffusion and Etest. Diagn. Microbiol. Infect. Dis. 1999, 34, 1–6. [Google Scholar] [CrossRef]
- Brazier, J.S.; Fawley, W.; Freeman, J.; Wilcox, M.H. Reduced susceptibility of Clostridium difficile to metronidazole. J. Antimicrob. Chemother. 2001, 48, 741–742. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, A.; Jackson, J.L.; Doi, A.; Kamiya, T. Metronidazole-induced central nervous system toxicity: A systematic review. Clin. Neuropharmacol. 2011, 34, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Pardi, D.S. Clostridium difficile infection: New insights into management. Mayo Clin. Proc. 2012, 87, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.J.; Emery, J.; Krulicki, W.; Byrne, B.; Mah, M. Opt-80 eliminates Clostridium difficile and is sparing of Bacteroides species during treatment of C. difficile infection. Antimicrob. Agents Chemother. 2009, 53, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Shue, Y.K.; Sears, P.S.; Shangle, S.; Walsh, R.B.; Lee, C.; Gorbach, S.L.; Okumu, F.; Preston, R.A. Safety, tolerance, and pharmacokinetic studies of Opt-80 in healthy volunteers following single and multiple oral doses. Antimicrob. Agents Chemother. 2008, 52, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Artsimovitch, I.; Seddon, J.; Sears, P. Fidaxomicin is an inhibitor of the initiation of bacterial RNA synthesis. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 55 (Suppl. 2), S127–S131. [Google Scholar] [CrossRef] [PubMed]
- Cornely, O.A.; Nathwani, D.; Ivanescu, C.; Odufowora-Sita, O.; Retsa, P.; Odeyemi, I.A.O. Clinical efficacy of fidaxomicin compared with vancomycin and metronidazole in Clostridium difficile infections: A meta-analysis and indirect treatment comparison. J. Antimicrob. Chemother. 2014, 69, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.J.; Cannon, K.; Byrne, B.; Emery, J.; Ward, L.; Eyben, M.; Krulicki, W. Fidaxomicin preserves the intestinal microbiome during and after treatment of Clostridium difficile infection (CDI) and reduces both toxin reexpression and recurrence of CDI. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2012, 55 (Suppl. 2), S132–S142. [Google Scholar] [CrossRef] [PubMed]
- Wösten, M.M. Eubacterial sigma-factors. FEMS Microbiol. Rev. 1998, 22, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.; Wilcox, M.H. Antibiotic activity against genotypically distinct and indistinguishable Clostridium difficile isolates. J. Antimicrob. Chemother. 2001, 47, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Zühlke, D.; Pané-Farré, J.; Kusch, H.; Wolf, C.; Reiß, S.; Binh, L.T.N.; Albrecht, D.; Riedel, K.; Hecker, M.; et al. Aureolib—A proteome signature library: Towards an understanding of Staphylococcus aureus pathophysiology. PLoS ONE 2013, 8, e70669. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Antelmann, H.; Büttner, K.; Bernhardt, J. Gel-based proteomics of Gram-positive bacteria: A powerful tool to address physiological questions. Proteomics 2008, 8, 4958–4975. [Google Scholar] [CrossRef] [PubMed]
- Bandow, J.E.; Brötz, H.; Leichert, L.I.O.; Labischinski, H.; Hecker, M. Proteomic approach to understanding antibiotic action. Antimicrob. Agents Chemother. 2003, 47, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.A.; Roberts, A.P.; Mullany, P. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916ΔE enters the genome of this strain at multiple sites. J. Med. Microbiol. 2005, 54, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, J.; Weibezahn, J.; Scharf, C.; Hecker, M. Bacillus subtilis during feast and famine: Visualization of the overall regulation of protein synthesis during glucose starvation by proteome analysis. Genome Res. 2003, 13, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Schwarz, K.; Fiedler, T.; Fischer, R.-J.; Bahl, H. A Standard Operating Procedure (SOP) for the preparation of intra- and extracellular proteins of Clostridium acetobutylicum for proteome analysis. J. Microbiol. Methods 2007, 68, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Moche, M.; Albrecht, D.; Maaß, S.; Hecker, M.; Westermeier, R.; Büttner, K. The new horizon in 2D electrophoresis-new technology to increase resolution and sensitivity. Electrophoresis 2013, 34, 1510–1518. [Google Scholar] [CrossRef] [PubMed]
- Dannheim, H.; Riedel, T.; Neumann-Schaal, M.; Bunk, B.; Schober, I.; Spröer, C.; Chibani, C.M.; Gronow, S.; Liesegang, H.; Overmann, J.; et al. Manual curation and reannotation of the genomes of Clostridium difficile 630Δerm and Clostridium difficile 630. J. Med. Microbiol. 2017, 66, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Dembek, M.; Barquist, L.; Boinett, C.J.; Cain, A.K.; Mayho, M.; Lawley, T.D.; Fairweather, N.F.; Fagan, R.P. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. mBio 2015, 6, e02383-14. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Bandow, J.E. Proteomic signatures in antibiotic research. Proteomics 2011, 11, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.C.; Citron, D.M.; Tyrrell, K.L.; Warren, Y.A. Bactericidal activity of telavancin, vancomycin and metronidazole against Clostridium difficile. Anaerobe 2010, 16, 220–222. [Google Scholar] [CrossRef] [PubMed]
- Haft, D.H.; Selengut, J.D.; White, O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003, 31, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Declerck, P.J.; De Ranter, C.J. In vitro reductive activation of nitroimidazoles. Biochem. Pharmacol. 1986, 35, 59–61. [Google Scholar] [CrossRef]
- Neumann-Schaal, M.; Hofmann, J.D.; Will, S.E.; Schomburg, D. Time-resolved amino acid uptake of Clostridium difficile 630Δerm and concomitant fermentation product and toxin formation. BMC Microbiol. 2015, 15, 281. [Google Scholar] [CrossRef] [PubMed]
- Maaß, S.; Wachlin, G.; Bernhardt, J.; Eymann, C.; Fromion, V.; Riedel, K.; Becher, D.; Hecker, M. Highly precise quantification of protein molecules per cell during stress and starvation responses in Bacillus subtilis. Mol. Cell. Proteomics 2014, 13, 2260–2276. [Google Scholar] [CrossRef]
- Hessling, B.; Bonn, F.; Otto, A.; Herbst, F.-A.; Rappen, G.-M.; Bernhardt, J.; Hecker, M.; Becher, D. Global proteome analysis of vancomycin stress in Staphylococcus aureus. Int. J. Med. Microbiol. 2013, 303, 624–634. [Google Scholar] [CrossRef] [PubMed]
- VanBogelen, R.A.; Neidhardt, F.C. Ribosomes as sensors of heat and cold shock in Escherichia coli. Proc. Natl. Acad. Sci. USA 1990, 87, 5589–5593. [Google Scholar] [CrossRef] [PubMed]
- Chong, P.M.; Lynch, T.; McCorrister, S.; Kibsey, P.; Miller, M.; Gravel, D.; Westmacott, G.R.; Mulvey, M.R. Canadian Nosocomial Infection Surveillance Program (CNISP). Proteomic analysis of a NAP1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS ONE 2014, 9, e82622. [Google Scholar] [CrossRef] [PubMed]
- Emerson, J.E.; Stabler, R.A.; Wren, B.W.; Fairweather, N.F. Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J. Med. Microbiol. 2008, 57, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Steffens, L.S.; Nicholson, S.; Paul, L.V.; Nord, C.E.; Patrick, S.; Abratt, V.R. Bacteroides fragilis RecA protein overexpression causes resistance to metronidazole. Res. Microbiol. 2010, 161, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Mukhopadhyay, A.K.; Dailidiene, D.; Wang, Y.; Velapatiño, B.; Gilman, R.H.; Parkinson, A.J.; Nair, G.B.; Wong, B.C.; Lam, S.K.; et al. Sequential inactivation of rdxA (HP0954) and frxA(HP0642) nitroreductase genes causes moderate and high-level metronidazole resistance in Helicobacter pylori. J. Bacteriol. 2000, 182, 5082–5090. [Google Scholar] [CrossRef] [PubMed]
- Gal, M.; Brazier, J.S. Metronidazole resistance in Bacteroides spp. carrying nim genes and the selection of slow-growing metronidazole-resistant mutants. J. Antimicrob. Chemother. 2004, 54, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Kwon, D.H.; Kato, M.; El-Zaatari, F.A.; Osato, M.S.; Graham, D.Y. Frame-shift mutations in NAD(P)H flavin oxidoreductase encoding gene (frxA) from metronidazole resistant Helicobacter pylori ATCC43504 and its involvement in metronidazole resistance. FEMS Microbiol. Lett. 2000, 188, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.E. Bacterial tellurite resistance. Trends Microbiol. 1999, 7, 111–115. [Google Scholar] [CrossRef]
- Chasteen, T.G.; Fuentes, D.E.; Tantaleán, J.C.; Vásquez, C.C. Tellurite: History, oxidative stress, and molecular mechanisms of resistance. FEMS Microbiol. Rev. 2009, 33, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Deng, W.; Yang, W.; Luo, H.; Duan, X.; Xie, L.; Li, P.; Wang, R.; Fu, T.; Abdalla, A.E.; et al. Mycobacterium tuberculosis Rv1152 is a novel GntR family transcriptional regulator involved in intrinsic vancomycin resistance and is a potential vancomycin adjuvant target. Sci. Rep. 2016, 6, 28002. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhao, L.; Yang, M. A GntR family transcription factor positively regulates mycobacterial isoniazid resistance by controlling the expression of a putative permease. BMC Microbiol. 2015, 15, 214. [Google Scholar] [CrossRef] [PubMed]
- White, D.G.; Goldman, J.D.; Demple, B.; Levy, S.B. Role of the acrAB locus in organic solvent tolerance mediated by expression of marA, soxS, or robA in Escherichia coli. J. Bacteriol. 1997, 179, 6122–6126. [Google Scholar] [CrossRef] [PubMed]
- Aono, R. Improvement of organic solvent tolerance level of Escherichia coli by overexpression of stress-responsive genes. Extremophiles 1998, 2, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Ariza, R.R.; Cohen, S.P.; Bachhawat, N.; Levy, S.B.; Demple, B. Repressor mutations in the marRAB operon that activate oxidative stress genes and multiple antibiotic resistance in Escherichia coli. J. Bacteriol. 1994, 176, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Moken, M.C.; McMurry, L.M.; Levy, S.B. Selection of multiple-antibiotic-resistant (mar) mutants of Escherichia coli by using the disinfectant pine oil: Roles of the mar and acrAB loci. Antimicrob. Agents Chemother. 1997, 41, 2770–2772. [Google Scholar] [CrossRef] [PubMed]
- McMurry, L.M.; Oethinger, M.; Levy, S.B. Overexpression of marA, soxS, or acrAB produces resistance to triclosan in laboratory and clinical strains of Escherichia coli. FEMS Microbiol. Lett. 1998, 166, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Okusu, H.; Ma, D.; Nikaido, H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 1996, 178, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.D.; White, D.G.; Levy, S.B. Multiple antibiotic resistance (mar) locus protects Escherichia coli from rapid cell killing by fluoroquinolones. Antimicrob. Agents Chemother. 1996, 40, 1266–1269. [Google Scholar] [CrossRef] [PubMed]
- Alekshun, M.N.; Levy, S.B. The mar regulon: Multiple resistance to antibiotics and other toxic chemicals. Trends Microbiol. 1999, 7, 410–413. [Google Scholar] [CrossRef]
- Spengler, G.; Kincses, A.; Gajdács, M.; Amaral, L. New roads leading to old destinations: Efflux pumps as targets to reverse multidrug resistance in bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession Number | Protein Name | R20291-Homologue | Function | Significantly Changed (p < 0.05, log2 Fold Change >|0.8|) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Amount | Synthesis | ||||||||
| VAN | MTZ | FDX | VAN | MTZ | FDX | ||||
| CDIF630erm_01408 | RimM | CDR20291_1095 | 16S rRNA processing protein RimM | x | x | x | |||
| CDIF630erm_03522 | DapB1 | CDR20291_3086 | 4-hydroxy-tetrahydrodipicolinate reductase | x | x | x | x | ||
| CDIF630erm_03707 | MurB | CDR20291_3224 | UDP-N-acetylenolpyruvoyl-glucosamine reductase | x | x | ||||
| CDIF630erm_02841 | RpoZ | CDR20291_2474 | DNA-directed RNA polymerase subunit omega | x | x | ||||
| CDIF630erm_01509 | CDIF630erm_01509 | CDR20291_1195 | putative pyridoxine kinase | x | |||||
| CDIF630erm_02877 | CDIF630erm_02877 | CDR20291_2507 | putative pyridoxal 5′-phosphate-dependent enzyme | x | |||||
| CDIF630erm_02708 | GrpE | CDR20291_2355 | stress protein (HSP-70 cofactor) | x | |||||
| CDIF630erm_04005 | MnmG | CDR20291_3535 | tRNA uridine 5-carboxymethyl-aminomethyl modification enzyme | x | |||||
| CDIF630erm_00239 | MurA | CDR20291_0122 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | x | |||||
| CDIF630erm_03828 | Prs | CDR20291_3351 | ribose-phosphate pyrophosphokinase | x | |||||
| CDIF630erm_00260 | SecA1 | CDR20291_0142 | protein translocase subunit SecA1 | x | |||||
| CDIF630erm_01292 | MreB2 | CDR20291_0982 | rod shape-determining protein MreB | x | x | ||||
| CDIF630erm_03832 | MurC | CDR20291_3355 | UDP-N-acetylmuramate-l-alanine ligase | x | x | ||||
| CDIF630erm_03585 | Pgi | CDR20291_3146 | glucose-6-phosphate isomerase | x | |||||
| CDIF630erm_02574 | GuaB | CDR20291_2224 | IMP dehydrogenase | x | |||||
| CDIF630erm_03992 | RpsF | CDR20291_3523 | 30S ribosomal protein S6 | x | |||||
| CDIF630erm_00160 | Adk | CDR20291_0088 | adenylate kinase | x | |||||
| CDIF630erm_02772 | LeuS | CDR20291_2410 | leucine-tRNA ligas | x | |||||
| CDIF630erm_03872 | LysS | CDR20291_3389 | lysine-tRNA ligase | x | |||||
| CDIF630erm_00461 | CDIF630erm_00461 | CDR20291_0338 | cobalt-dependent inorganic pyrophosphatase | x | |||||
| Antibiotic | Accession Number | Protein Name | Function | Regulation |
|---|---|---|---|---|
| VAN | CDIF630erm_00928 | FloX | putative flavodoxin | ↑ |
| VAN | CDIF630erm_00005 | GyrB | DNA gyrase subunit B | ↑ |
| VAN | CDIF630erm_01147 | NagB | glucosamine-6-phosphate deaminase | ↑ |
| VAN | CDIF630erm_03585 | Pgi | glucose-6-phosphate isomeras | ↑ |
| VAN | CDIF630erm_02205 | CspC1 | major cold shock protein CspC | ↓ |
| VAN | CDIF630erm_03462 | Eno | enolase | ↓ |
| VAN | CDIF630erm_01010 | SpeE | spermidine synthase | ↓ |
| VAN | CDIF630erm_03837 | RsmA | ribosomal RNA small subunit methyltransferase A | ↑↓ |
| MTZ | CDIF630erm_03239 | AtpA1 | V-type ATP synthase subunit A | ↑ |
| MTZ | CDIF630erm_03421 | BglA5 | 6-phospho-beta-glucosidase | ↑ |
| MTZ | CDIF630erm_03910 | CarB1 | carbamoyl-phosphate synthase large subunit | ↑ |
| MTZ | CDIF630erm_03912 | CarB2 | carbamoyl-phosphate synthase large subunit | ↑ |
| MTZ | CDIF630erm_00291 | CDIF630erm_00291 | ABC-type transport system, ATP-binding protein | ↑ |
| MTZ | CDIF630erm_02352 | CDIF630erm_02352 | putative multiprotein-complex assembly TPR repeat-containing protein | ↑ |
| MTZ | CDIF630erm_03387 | CDIF630erm_03387 | PTS system, glucose-like IIA component | ↑ |
| MTZ | CDIF630erm_03611 | CDIF630erm_03611 | putative D-isomer specific 2-hydroxyacid dehydrogenase | ↑ |
| MTZ | CDIF630erm_03703 | CDIF630erm_03703 | putative hydrolase, NUDIX family | ↑ |
| MTZ | CDIF630erm_03788 | ComEB2 | deoxycytidylate deaminase | ↑ |
| MTZ | CDIF630erm_01952 | Def1 | peptide deformylase | ↑ |
| MTZ | CDIF630erm_02427 | FtnA | bacterial non-heme ferritin | ↑ |
| MTZ | CDIF630erm_03003 | HisS | histidine-tRNA ligase | ↑ |
| MTZ | CDIF630erm_01131 | Hpr | hydroxypyruvate reductase | ↑ |
| MTZ | CDIF630erm_02713 | LepA | elongation factor 4 | ↑ |
| MTZ | CDIF630erm_02845 | LtaE | low specificity l-threonine aldolase | ↑ |
| MTZ | CDIF630erm_04005 | MnmG | tRNA uridine 5-carboxymethylaminomethyl modification enzyme | ↑ |
| MTZ | CDIF630erm_01275 | PolA | DNA polymerase I (POLI) | ↑ |
| MTZ | CDIF630erm_01160 | PotA | ABC-type transport system, spermidine/putrescine ATP-binding protein | ↑ |
| MTZ | CDIF630erm_00260 | SecA1 | protein translocase subunit SecA1 | ↑ |
| MTZ | CDIF630erm_01481 | SelU | tRNA 2-selenouridine synthase | ↑ |
| MTZ | CDIF630erm_01811 | TerD1 | tellurium resistance protein TerD | ↑ |
| MTZ | CDIF630erm_02559 | Tkt’1 | transketolase, C-terminal section | ↑ |
| MTZ | CDIF630erm_00773 | ToxB | toxin B | ↑ |
| MTZ | CDIF630erm_01943 | YjiM | putative 2-hydroxyacyl-CoA dehydratase | ↑ |
| MTZ | CDIF630erm_01323 | AckA | acetate kinase | ↓ |
| MTZ | CDIF630erm_02495 | AmpP | xaa-pro aminopeptidase | ↓ |
| MTZ | CDIF630erm_02343 | BipA | GTP-binding protein BipA | ↓ |
| MTZ | CDIF630erm_01509 | CDIF630erm_01509 | putative pyridoxine kinase | ↓ |
| MTZ | CDIF630erm_01961 | CDIF630erm_01961 | putative phenylalanyl-tRNA synthetase beta chain | ↓ |
| MTZ | CDIF630erm_02609 | CDIF630erm_02609 | DsrEFH-like protein | ↓ |
| MTZ | CDIF630erm_02799 | CDIF630erm_02799 | hypothetical protein | ↓ |
| MTZ | CDIF630erm_02825 | CDIF630erm_02825 | putative nitroreductase-like oxidoreductase | ↓ |
| MTZ | CDIF630erm_02877 | CDIF630erm_02877 | putative pyridoxal 5′-phosphate-dependent enzyme | ↓ |
| MTZ | CDIF630erm_02881 | CDIF630erm_02881 | ACT domain-containing protein | ↓ |
| MTZ | CDIF630erm_03134 | CDIF630erm_03134 | putative modulator of DNA gyrase, peptidase U62 | ↓ |
| MTZ | CDIF630erm_03311 | CDIF630erm_03311 | PTS system, arabinose-specific IIA component | ↓ |
| MTZ | CDIF630erm_02708 | GrpE | protein (HSP-70 cofactor) | ↓ |
| MTZ | CDIF630erm_02574 | GuaB | IMP dehydrogenase | ↓ |
| MTZ | CDIF630erm_03527 | Hpt2 | hypoxanthine phosphoribosyltransferase | ↓ |
| MTZ | CDIF630erm_00239 | MurA | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | ↓ |
| MTZ | CDIF630erm_00113 | ProS2 | proline-tRNA ligase | ↓ |
| MTZ | CDIF630erm_03828 | Prs | ribose-phosphate pyrophosphokinase | ↓ |
| MTZ | CDIF630erm_03992 | RpsF | 30S ribosomal protein S6 | ↓ |
| MTZ | CDIF630erm_00009 | RsbV | anti-sigma factor antagonist | ↓ |
| MTZ | CDIF630erm_03996 | YedF | selenium metabolism protein YedF | ↓ |
| MTZ | CDIF630erm_01998 | TerD4 | tellurium resistance protein TerD | ↑↓ |
| FDX | CDIF630erm_00843 | AcsE | 5-methyltetrahydrofolate|corrinoid/iron-sulfur protein co-methyltransferase | ↑ |
| FDX | CDIF630erm_03250 | AdhE2 | aldehyde-alcohol dehydrogenase | ↑ |
| FDX | CDIF630erm_00160 | Adk | adenylate kinase | ↑ |
| FDX | CDIF630erm_00657 | ArcB | Delta (1)-pyrroline-2-carboxylate reductase | ↑ |
| FDX | CDIF630erm_00363 | CDIF630erm_00363 | phosphothreonine phosphatase | ↑ |
| FDX | CDIF630erm_00461 | CDIF630erm_00461 | cobalt-dependent inorganic pyrophosphatase | ↑ |
| FDX | CDIF630erm_01443 | CDIF630erm_01443 | ribonuclease J family protein | ↑ |
| FDX | CDIF630erm_01543 | CDIF630erm_01543 | transcriptional regulator, GntR family | ↑ |
| FDX | CDIF630erm_01816 | CDIF630erm_01816 | putative tellurite associated resistance protein | ↑ |
| FDX | CDIF630erm_01906 | CDIF630erm_01906 | uncharacterized protein | ↑ |
| FDX | CDIF630erm_02819 | CDIF630erm_02819 | PTS system, mannosylglycerate-specific IIA component | ↑ |
| FDX | CDIF630erm_03499 | CDIF630erm_03499 | putative nitroreductase-like oxidoreductase | ↑ |
| FDX | CDIF630erm_03853 | CDIF630erm_03853 | putative deoxyribonuclease | ↑ |
| FDX | CDIF630erm_00963 | Cwp25 | putative cell wall-binding protein | ↑ |
| FDX | CDIF630erm_01328 | FabH | beta-ketoacyl-[acyl-carrier-protein] synthase 3 | ↑ |
| FDX | CDIF630erm_00361 | FliC | flagellin C | ↑ |
| FDX | CDIF630erm_00114 | GltX | glutamate-tRNA ligase | ↑ |
| FDX | CDIF630erm_02400 | Hcp | hydroxylamine reductase | ↑ |
| FDX | CDIF630erm_03727 | HemB | delta-aminolevulinic acid dehydratase | ↑ |
| FDX | CDIF630erm_02772 | LeuS | leucine-tRNA ligase | ↑ |
| FDX | CDIF630erm_03872 | LysS | lysine-tRNA ligase | ↑ |
| FDX | CDIF630erm_03918 | MetH | methionine synthase | ↑ |
| FDX | CDIF630erm_01292 | MreB2 | rod shape-determining protein MreB | ↑ |
| FDX | CDIF630erm_01369 | NudF | ADP-ribose pyrophosphatase | ↑ |
| FDX | CDIF630erm_00345 | PurH | bifunctional phosphoribosylaminoimidazolecarboxamide formyltransferase/IMP cyclohydrolase | ↑ |
| FDX | CDIF630erm_02743 | SelD | selenide, water dikinase | ↑ |
| FDX | CDIF630erm_02763 | TdcB | threonine dehydratase | ↑ |
| FDX | CDIF630erm_02348 | ThrB | homoserine kinase | ↑ |
| FDX | CDIF630erm_03374 | TreA | trehalose-6-phosphate hydrolase | ↑ |
| FDX | CDIF630erm_00231 | VorB1 | 3-methyl-2-oxobutanoate dehydrogenase (ferredoxin), beta subunit | ↑ |
| FDX | CDIF630erm_02749 | YcsE | putative 5-amino-6-(5-phospho-d-ribitylamino) uracil phosphatase | ↑ |
| FDX | CDIF630erm_03774 | Alr | alanine racemase | ↓ |
| FDX | CDIF630erm_01216 | ApeA | aminopeptidase ApeA | ↓ |
| FDX | CDIF630erm_00601 | CDIF630erm_00601 | transcriptional regulator, MarR family | ↓ |
| FDX | CDIF630erm_00852 | CDIF630erm_00852 | uncharacterized protein | ↓ |
| FDX | CDIF630erm_01899 | CDIF630erm_01899 | uncharacterized protein, UPF0145 family | ↓ |
| FDX | CDIF630erm_02209 | CDIF630erm_02209 | uncharacterized protein, AhpD-like | ↓ |
| FDX | CDIF630erm_02775 | CDIF630erm_02775 | putative hydrolase | ↓ |
| FDX | CDIF630erm_00360 | FliT | flagellar protein FliT | ↓ |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maaß, S.; Otto, A.; Albrecht, D.; Riedel, K.; Trautwein-Schult, A.; Becher, D. Proteomic Signatures of Clostridium difficile Stressed with Metronidazole, Vancomycin, or Fidaxomicin. Cells 2018, 7, 213. https://doi.org/10.3390/cells7110213
Maaß S, Otto A, Albrecht D, Riedel K, Trautwein-Schult A, Becher D. Proteomic Signatures of Clostridium difficile Stressed with Metronidazole, Vancomycin, or Fidaxomicin. Cells. 2018; 7(11):213. https://doi.org/10.3390/cells7110213
Chicago/Turabian StyleMaaß, Sandra, Andreas Otto, Dirk Albrecht, Katharina Riedel, Anke Trautwein-Schult, and Dörte Becher. 2018. "Proteomic Signatures of Clostridium difficile Stressed with Metronidazole, Vancomycin, or Fidaxomicin" Cells 7, no. 11: 213. https://doi.org/10.3390/cells7110213
APA StyleMaaß, S., Otto, A., Albrecht, D., Riedel, K., Trautwein-Schult, A., & Becher, D. (2018). Proteomic Signatures of Clostridium difficile Stressed with Metronidazole, Vancomycin, or Fidaxomicin. Cells, 7(11), 213. https://doi.org/10.3390/cells7110213