Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors


Abstract
:1. Introduction
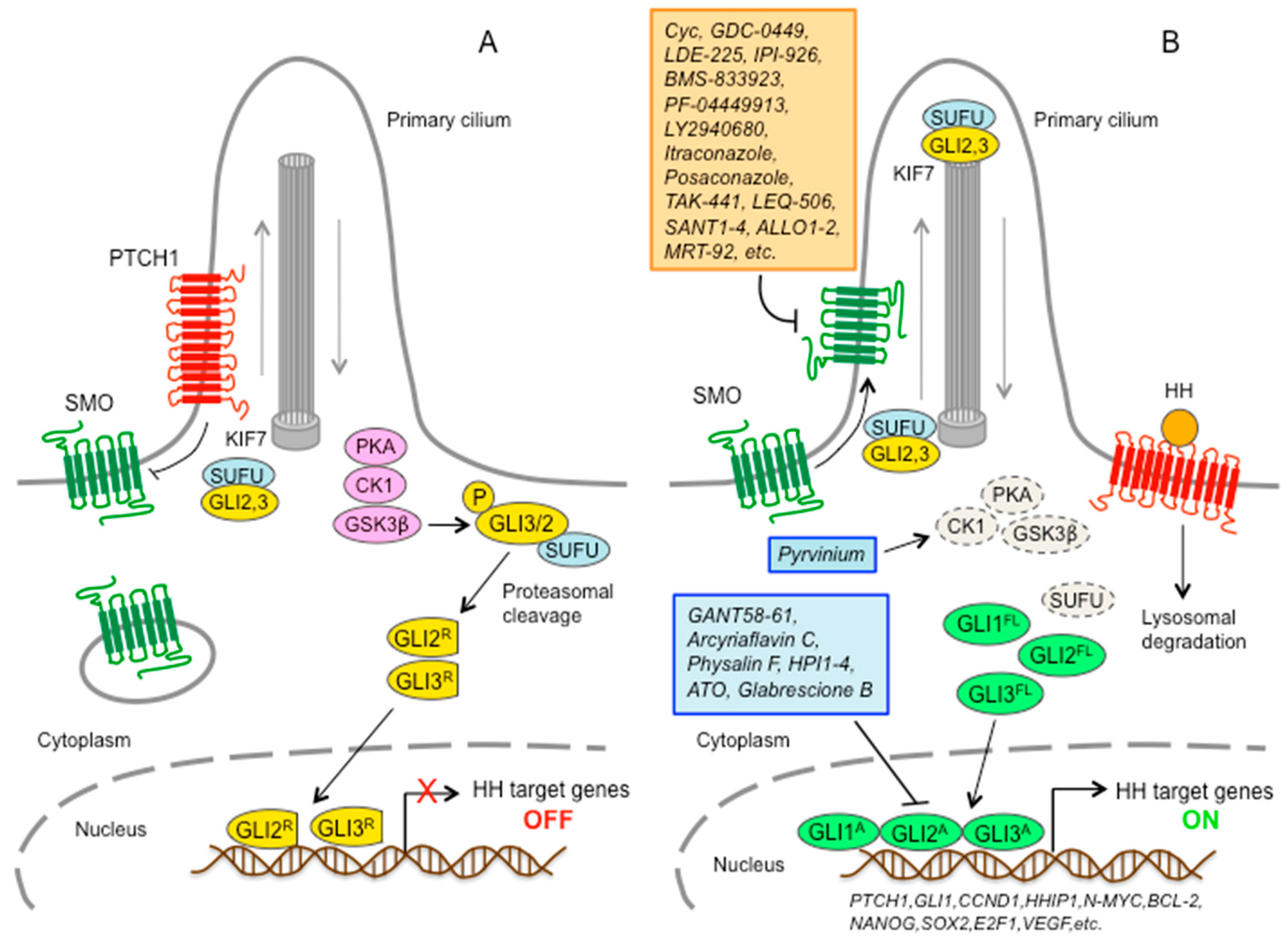
2. Hedgehog-GLI Signaling Pathway at a Glance
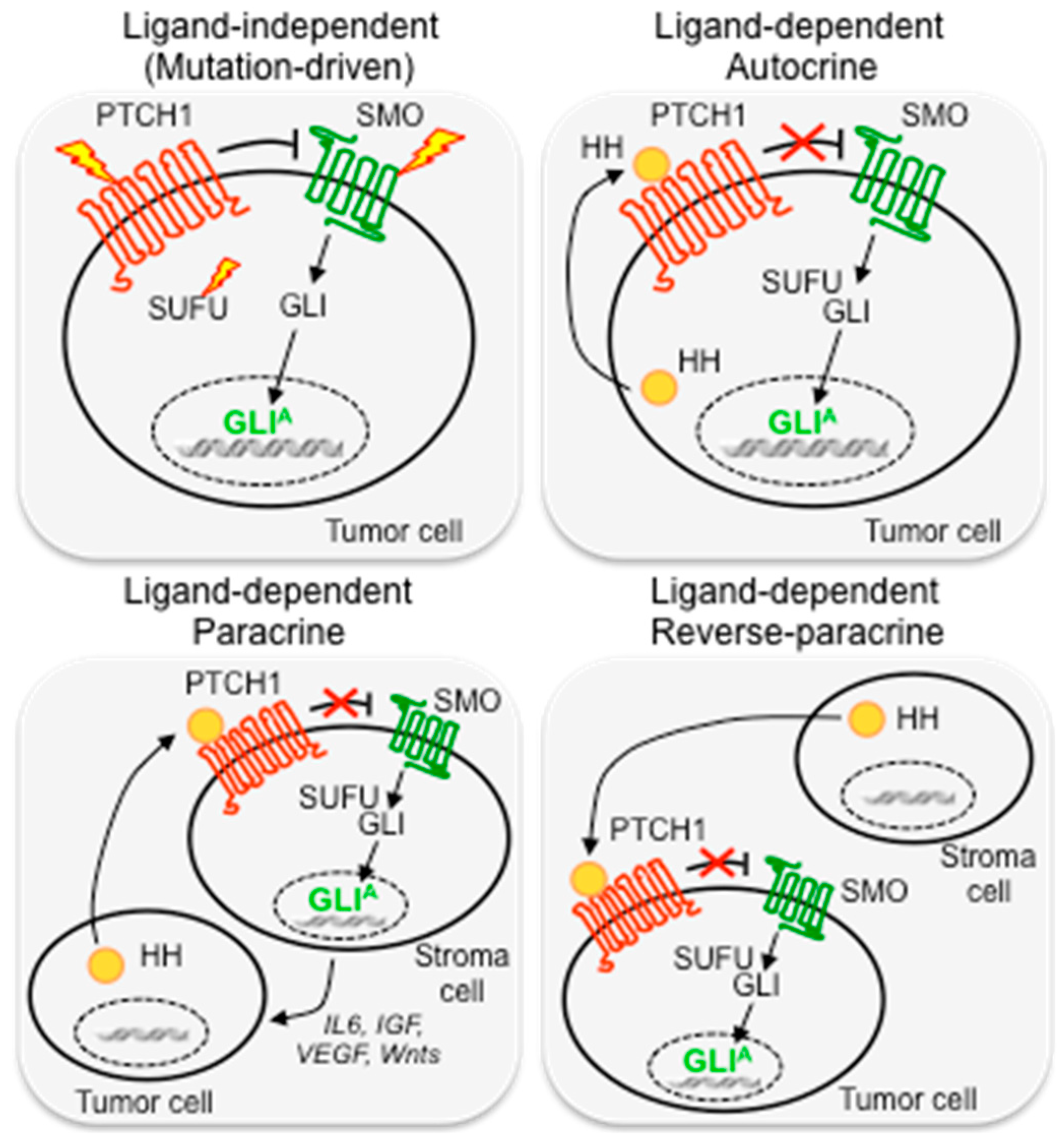
Mechanisms of Hedgehog Pathway Activation in Cancer
3. Smoothened: Structure of the Receptor and Mutations in Cancer
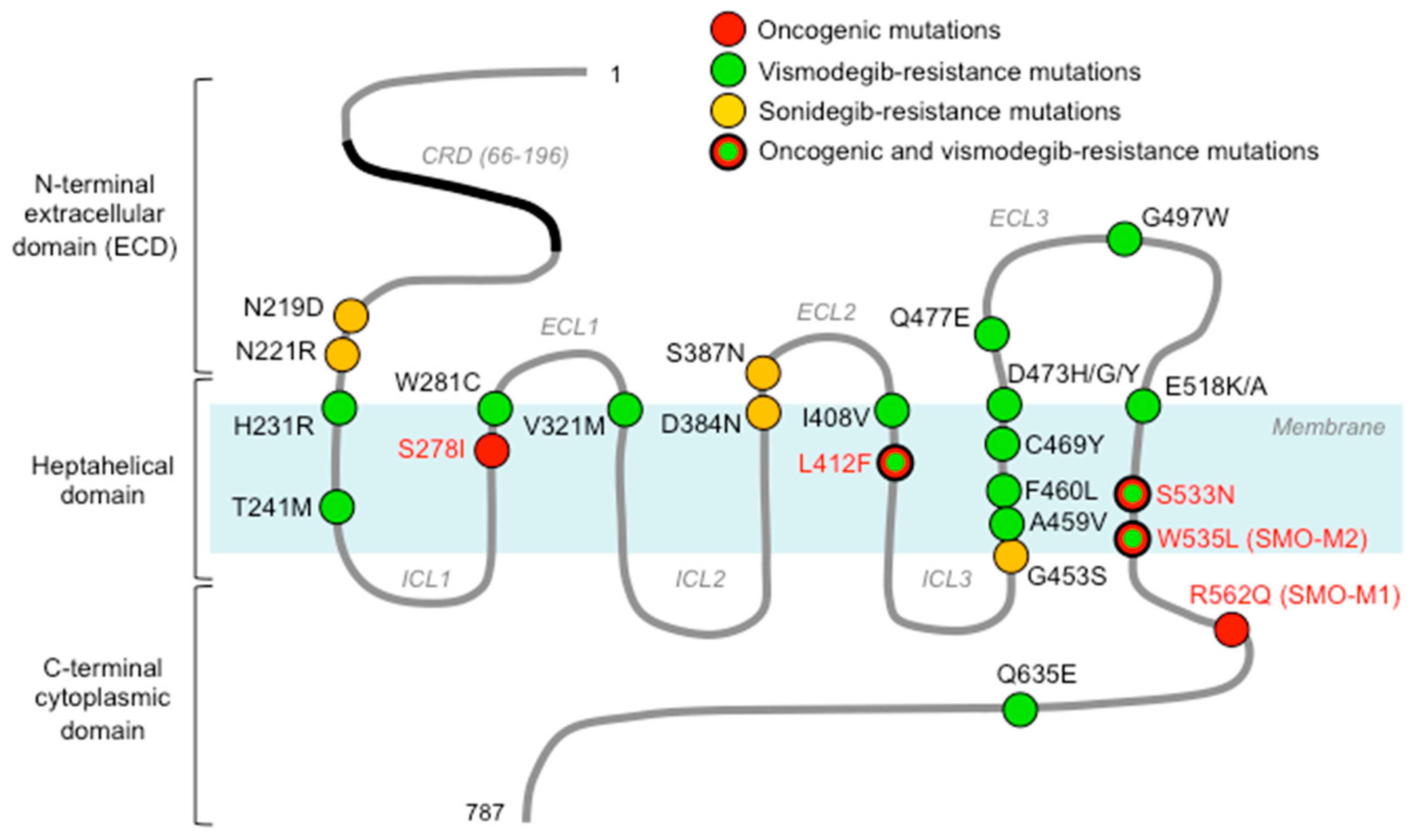
3.1. Structure of Smoothened
3.2. Smoothened Binding Sites
3.3. Oncogenic Smoothened Mutations
4. Smoothened Inhibitors
4.1. SMO Inhibitors in Clinical Trials
4.1.1. Vismodegib (GDC-0449)
4.1.2. Sonidegib (Erismodegib, LDE-225, NVP-LDE225)
4.1.3. Saridegib (IPI-926)
4.1.4. BMS-833923 (XL139)
4.1.5. PF-04449913 (Glasdegib)
4.1.6. LY2940680 (Taladegib)
4.1.7. TAK-441
4.1.8. LEQ-506
4.1.9. Vitamin D3
4.2. SMO Inhibitors in Preclinical Studies
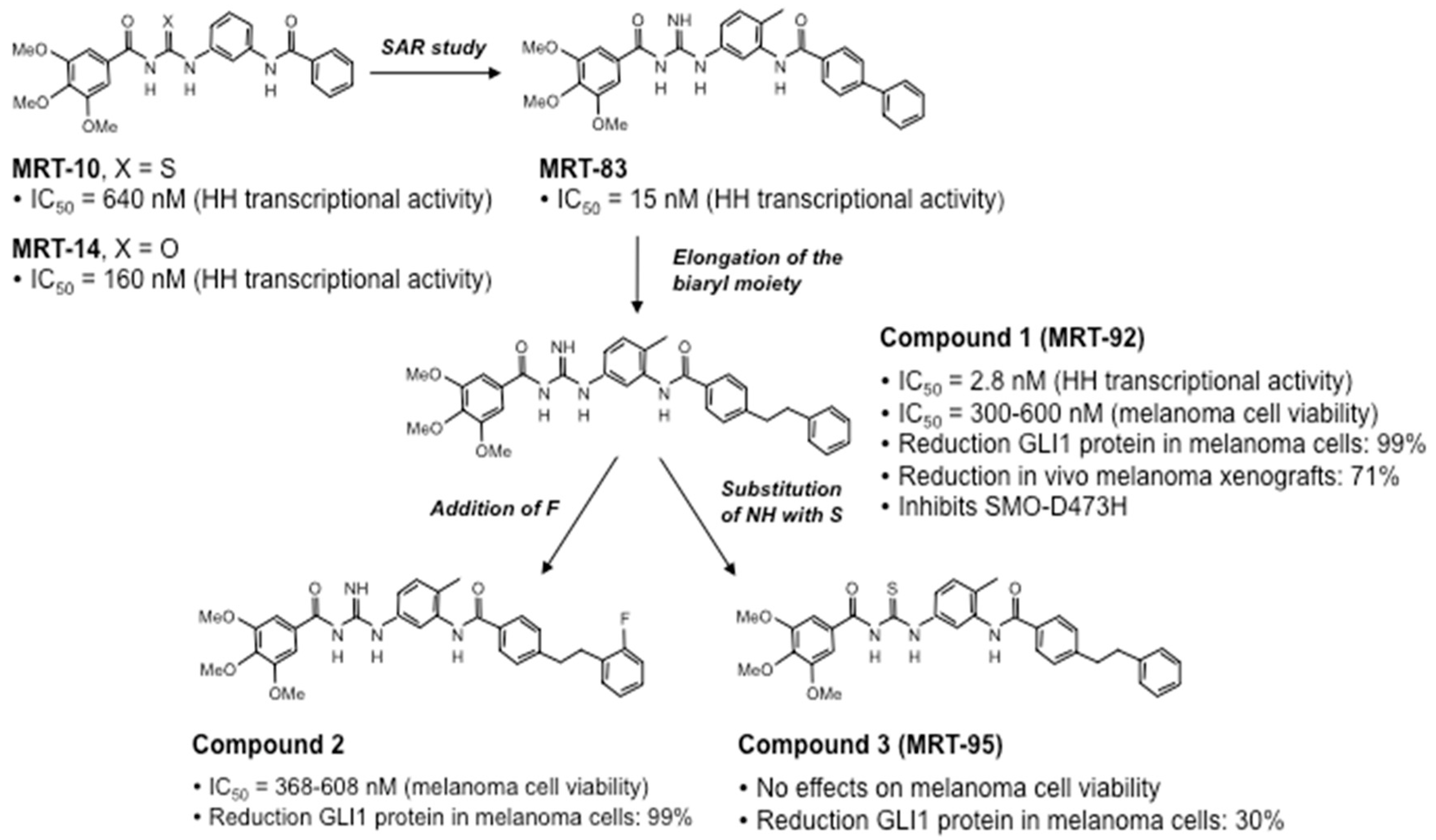
4.3. Novel Acylguanidine Derivatives as Potent SMO Antagonists
5. GLI Inhibitors
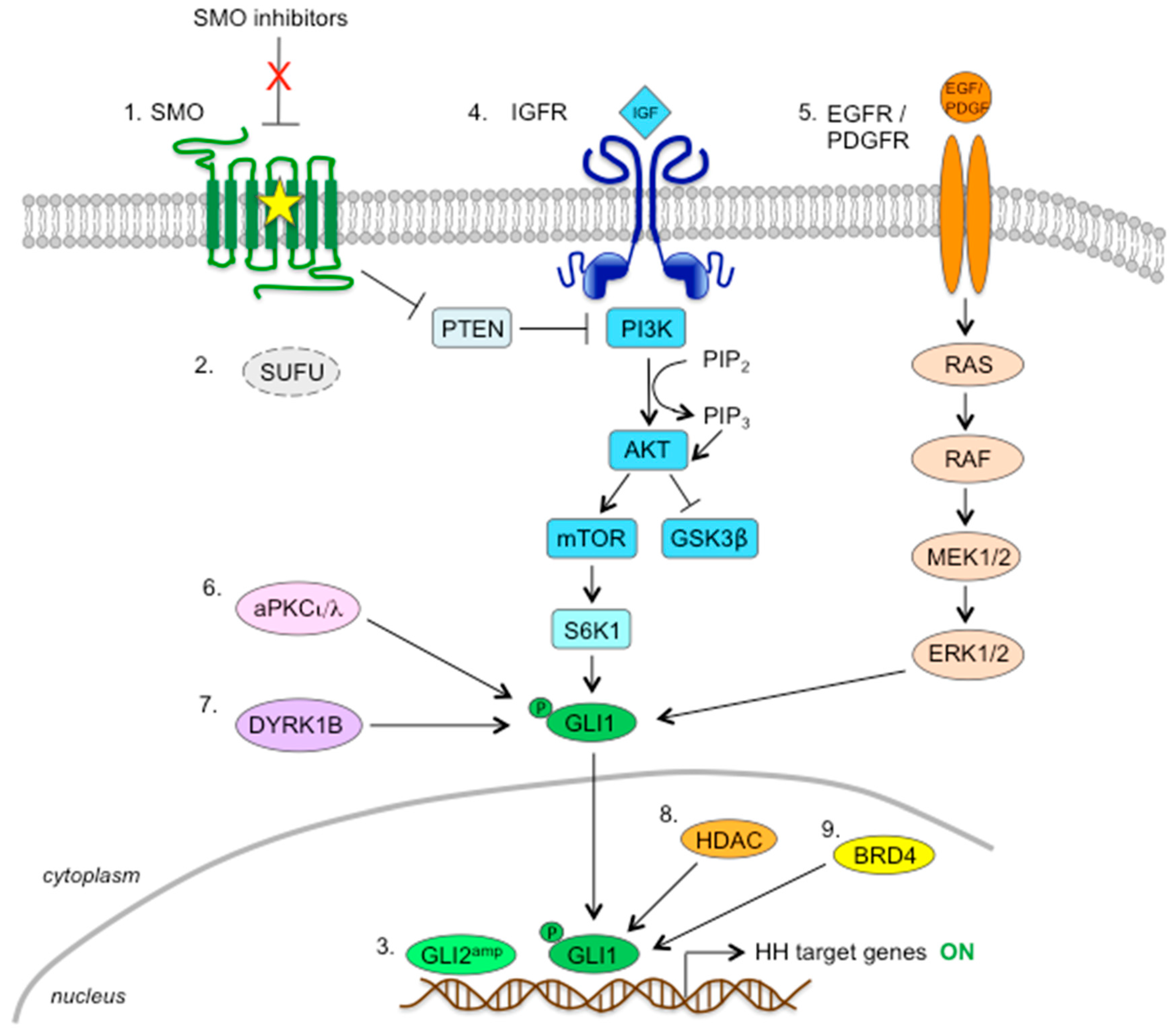
6. Mechanisms of Resistance to SMO Inhibitors
6.1. Resistance-Associated Smoothened Mutations
6.2. Activation of HH Pathway Downstream of SMO
6.3. Noncanonical, Compensatory Oncogenic Signaling Pathways
6.3.1. PI3K/AKT/mTOR Pathway
6.3.2. RAS-RAF-MEK-ERK Pathway
6.3.3. Protein Kinases
6.3.4. Chromatin Modulators
6.3.5. Other Mechanisms
7. Concluding Remarks
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
Abbreviations
| HH | Hedgehog-GLI |
| SMO | Smoothened |
| BCC | Basal cell carcinoma |
| MB | Medulloblastoma |
| PNET | Primitive neuroectodermal tumors |
| AML | Acute myeloid leukemia |
| SHH | Sonic hedgehog |
| IHH | Indian hedgehog |
| DHH | Desert hedgehog |
| PTCH1 | Patched 1 |
| GPCR | G-protein-coupled receptor |
| SUFU | Suppressor of Fused |
| KIF7 | Kinesin family member 7 |
| PC | Primary cilium |
| PKA | Protein kinase A |
| CK1 | Casein kinase 1 |
| GSK3β | Glycogen synthase kinase 3β |
| HHIP1 | Hedgehog-interacting protein |
| TNF-α | Tumor necrosis factor-α |
| mTOR | Mammalian target of rapamycin |
| S6K1 | S6 kinase 1 |
| ECD | Extracellular domain |
| CRD | Cysteine-rich domain |
| TMD | Transmembrane domain |
| ICL | Intracellular loop |
| ECL | Extracellular loop |
| Gprk2 | G protein-coupled receptor kinase 2 |
| PI4P | Phosphatidylinositol 4-phosphate |
| Cul4 | Cullin4 |
| DDB1 | DNA damage binding protein 1 |
| DBP | Drug-binding pocket |
| FDA | Food and Drug Administration |
| Pgp | P-glycoprotein |
| ATO | Arsenic trioxide |
| IGFR | Insulin growth factor receptor |
| PTEN | Phosphatase and tensin homolog |
| PI3K | Phosphatidylinositol-4,5-bisphosphate 3-kinase |
| EGFR | Epidermal growth factor receptor |
| PDGFR | Platelet-derived growth factor receptor |
| MAPK | Mitogen-Activated Protein kinase |
| ERK | Extracellular Signal-Regulated Kinase |
| aPKCι/λ | Atypical protein kinase Cι/λ |
| DYRK | Dual specificity tyrosine-phosphorylation-regulated kinase |
| HDAC | Histone deacetylase |
| BET | Bromo- and extra-terminal domain |
| SRF | Serum response factor |
| MLK1 | Megakaryoblastic leukemia 1 |
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Aanstad, P.; Singla, V.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Y. Evidence for the direct involvement of {beta}TrCP in Gli3 protein processing. Proc. Natl. Acad. Sci. USA 2006, 103, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, B. A novel protein-processing domain in Gli2 and Gli3 differentially blocks complete protein degradation by the proteasome. J. Biol. Chem. 2007, 282, 10846–10852. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol. Cell. Biol. 1990, 10, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Winklmayr, M.; Schmid, C.; Laner-Plamberger, S.; Kaser, A.; Aberger, F.; Eichberger, T.; Frischauf, A.M. Non-consensus GLI binding sites in Hedgehog target gene regulation. BMC Mol. Biol. 2010, 11, 2. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, V.; Benelli, M.; Apollo, A.; Pescucci, C.; Licastro, D.; Urso, C.; Gerlini, G.; Borgognoni, L.; Luzzatto, L.; Stecca, B. Thin and thick primary cutaneous melanomas reveal distinct patterns of somatic copy number alterations. Oncotarget 2016, 7, 30365–30378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernández, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz i Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Hegde, G.V.; Munger, C.M.; Emanuel, K.; Joshi, A.D.; Greiner, T.C.; Weisenburger, D.D.; Vose, J.M.; Joshi, S.S. Targeting of sonic hedgehog-GLI signaling: A potential strategy to improve therapy for mantle cell lymphoma. Mol. Cancer Ther. 2008, 7, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Montagnani, V.; Penachioni, J.Y.; Vinci, M.C.; Olivito, B.; Borgognoni, L.; Stecca, B. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 2013, 32, 4737–4747. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagani, Z.; Mora-Blanco, E.L.; Sansam, C.G.; McKenna, E.S.; Wilson, B.; Chen, D.; Klekota, J.; Tamayo, P.; Nguyen, P.T.; Tolstorukov, M.; et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat. Med. 2010, 16, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredriksson, R.; Lagerstrom, M.C.; Lundin, L.G.; Schioth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, F.; Wu, Y.; Yang, J.; Han, G.W.; Zhao, S.; Ishchenko, A.; Ye, L.; Lin, X.; Ding, K.; et al. Crystal structure of a multi-domain human smoothened receptor in complex with a super stabilizing ligand. Nat. Commun. 2017, 8, 15383. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Katritch, V.; Han, G.W.; Huang, X.P.; Liu, W.; Siu, F.Y.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Structure of the human smoothened receptor bound to an antitumour agent. Nature 2013, 497, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpel, S.; Claret, S.; Sanial, M.; Brigui, A.; Piolot, T.; Daviet, L.; Martin-Lannerée, S.; Plessis, A. The last 59 amino acids of Smoothened cytoplasmic tail directly bind the protein kinase Fused and negatively regulate the Hedgehog pathway. Dev. Biol. 2007, 303, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Williams, E.H.; Guo, Y.; Lum, L.; Beachy, P.A. Extensive phosphorylation of Smoothened in Hedgehog pathway activation. Proc. Natl. Acad. Sci. USA 2004, 101, 17900–17907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Ren, X.R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of Smoothened mediated by β-arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Maier, D.; Cheng, S.; Faubert, D.; Hipfner, D.R. A broadly conserved g-protein-coupled receptor kinase phosphorylation mechanism controls Drosophila smoothened activity. PLoS Genet. 2014, 10, e1004399. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Liu, Y.; Fan, J.; Zhang, J.; Li, X.A.; Evers, B.M.; Zhu, H.; Jia, J. PI(4)P Promotes Phosphorylation and Conformational Change of Smoothened through Interaction with Its C-terminal Tail. PLoS Biol. 2016, 14, e1002375. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tong, C.; Jiang, J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 2007, 450, 252–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, J.; Wright, S.C.; Rodríguez, D.; Matricon, P.; Lahav, N.; Vromen, A.; Friedler, A.; Strömqvist, J.; Wennmalm, S.; Carlsson, J.; et al. Agonist-induced dimer dissociation as a macromolecular step in G protein-coupled receptor signaling. Nat. Commun. 2017, 8, 226. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Nystedt, S.; Shivdasani, A.A.; Strutt, H.; Thomas, C.; Ingham, P.W. Functional domains and sub-cellular distribution of the Hedgehog transducing protein Smoothened in Drosophila. Mech. Dev. 2004, 121, 507–518. [Google Scholar] [CrossRef] [PubMed]
- DeCamp, D.L.; Thompson, T.M.; de Sauvage, F.J.; Lerner, M.R. Smoothened activates Galphai-mediated signalling in frog melanophores. J. Biol. Chem. 2000, 275, 26322–26327. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K.; Takahashi, M.; Osumi, N.; Sinnarajah, S.; Takeo, T.; Ikeda, H.; Kehrl, J.H.; Itoh, G.; Arnheiter, H. The G12 family of heterotrimeric G proteins and Rho GTPase mediate Sonic hedgehog signaling. Genes Cells 2004, 9, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Saucy, B.; Dilizio, C.; Manning, D.R. Activation of heterotrimeric G proteins by Smoothened. Proc. Natl. Acad. Sci. USA 2006, 103, 12607–12612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Cho, Y.S.; Wang, B.; Li, S.; Jiang, J. Regulation of Smoothened ubiquitylation and cell surface expression through a Cul4-DDB1-Gβ E3 ubiquitin ligase complex. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.E.; Marada, S.; Stewart, D.P.; Ouyang, J.X.; Ogden, S.K. The extracellular loops of Smoothened play a regulatory role in control of Hedgehog pathway activation. Development 2012, 139, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Kawasaki, T.; Mine, S.; Matsumura, H. Functional Role of the C-Terminal Amphipathic Helix 8 of Olfactory Receptors and Other G Protein-Coupled Receptors. Int. J. Mol. Sci. 2016, 17, 1930. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Zheng, S.; Wierbowski, B.M.; Kim, Y.; Nedelcu, D.; Aravena, L.; Liu, J.; Kruse, A.C.; Salic, A. Structural Basis of Smoothened Activation in Hedgehog Signaling. Cell 2018, 174, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wu, H.; Evron, T.; Vardy, E.; Han, G.W.; Huang, X.P.; Hufeisen, S.J.; Mangano, T.J.; Urban, D.J.; Katritch, V.; et al. Structural basis for Smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014, 5, 4355. [Google Scholar] [CrossRef] [PubMed]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Young, K.E.; Maiti, T.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank-Kamenetsky, M.; Zhang, X.M.; Bottega, S.; Guicherit, O.; Wichterle, H.; Dudek, H.; Bumcrot, D.; Wang, F.Y.; Jones, S.; Shulok, J.; et al. Small-molecule modulators of Hedgehog signaling: Identification and characterization of Smoothened agonists and antagonists. J. Biol. 2002, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Chen, J.K. Purmorphamine activates the Hedgehog pathway by targeting Smoothened. Nat. Chem. Biol. 2006, 2, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrne, E.F.X.; Sircar, R.; Miller, P.S.; Hedger, G.; Luchetti, G.; Nachtergaele, S.; Tully, M.D.; Mydock-McGrane, L.; Covey, D.F.; Rambo, R.P.; et al. Structural basis of Smoothened regulation by its extracellular domains. Nature 2016, 535, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. Chem. Med. Chem. 2013, 8, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Bender, M.H.; Hipskind, P.A.; Capen, A.R.; Cockman, M.; Credille, K.M.; Gao, H.; Bastian, J.A.; Clay, J.M.; Lobb, K.L.; Sall, D.J.; et al. Identification and characterization of a novel smoothened antagonist for the treatment of cancer with deregulated hedgehog signaling. Cancer Res. 2011, 71. [Google Scholar] [CrossRef]
- Nachtergaele, S.; Mydock, L.K.; Krishnan, K.; Rammohan, J.; Schlesinger, P.H.; Covey, D.F.; Rohatgi, R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat. Chem. Biol. 2012, 8, 211–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nachtergaele, S.; Whalen, D.M.; Mydock, L.K.; Zhao, Z.; Malinauskas, T.; Krishnan, K.; Ingham, P.W.; Covey, D.F.; Siebold, C.; Rohatgi, R. Structure and function of the Smoothened extracellular domain in vertebrate Hedgehog signaling. Elife 2013, 2, e01340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Rana, R.; Carroll, C.E.; Lee, H.J.; Bao, J.; Marada, S.; Grace, C.R.; Guibao, C.D.; Ogden, S.K.; Zheng, J.J. Structural insights into the role of the Smoothened cysteine-rich domain in Hedgehog signalling. Nat. Commun. 2013, 4, 2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijlsma, M.F.; Spek, C.A.; Zivkovic, D.; van de Water, S.; Rezaee, F.; Peppelenbosch, M.P. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006, 4, e232. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Cherezov, V.; Griffith, M.T.; Roth, C.B.; Jaakola, V.P.; Chien, E.Y.; Velasquez, J.; Kuhn, P.; Stevens, R.C. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure 2008, 16, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.P.; Griffith, M.T.; Hanson, M.A.; Cherezov, V.; Chien, E.Y.; Lane, J.R.; Ijzerman, A.P.; Stevens, R.C. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science 2008, 322, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Mathiesen, J.M.; Sunahara, R.K.; Pardo, L.; Weis, W.I.; Kobilka, B.K.; Granier, S. Crystal structure of the µ-opioid receptor bound to a morphinan antagonist. Nature 2012, 485, 321–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, B.R.; Sever, N.; Chong, Y.C.; Kim, J.; Belani, J.D.; Rychnovsky, S.; Bazan, J.F.; Beachy, P.A. Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev. Cell 2013, 26, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, R.T.; McClary, A.C.; Myers, B.R.; Biscocho, J.; Neahring, L.; Kwei, K.A.; Qu, K.; Gong, X.; Ng, T.; Jones, C.D.; et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat. Genet. 2014, 46, 722–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, KL.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Li, J.R.; Yao, C.Y.; Urman, N.M.; Chang, A.L.S.; Tang, J.Y.; Oro, A.E. Rolling the Genetic Dice: Neutral and Deleterious Smoothened Mutations in Drug-Resistant Basal Cell Carcinoma. J. Investig. Dermatol. 2015, 135, 2138–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pricl, S.; Cortelazzi, B.; Dal Col, V.; Marson, D.; Laurini, E.; Fermeglia, M.; Licitra, L.; Pilotti, S.; Bossi, P.; Perrone, F. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Mol. Oncol. 2015, 9, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keeler, R.F. Teratogenic effects of cyclopamine and jervine in rats, mice and hamsters. Proc. Soc. Exp. Biol. Med. 1975, 149, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Fendrich, V.; McGovern, K.; Bedja, D.; Bisht, S.; Alvarez, H.; Koorstra, J.B.; Habbe, N.; Karikari, C.; Mullendore, M.; et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol. Cancer Ther. 2008, 7, 2725–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Hann, C.L.; Laterra, J.; Yauch, R.L.; Callahan, C.A.; Fu, L.; Holcomb, T.; Stinson, J.; Gould, S.E.; Coleman, B.; et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 2009, 361, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a Potent and Selective Smoothened Antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef] [PubMed]
- Gendreau, S.B.; Hawkins, D.; Ho, C.P.; Lewin, A.; Lin, T.; Merchant, A.; Rowley, R.B.; Wang, Q.; Matsui, W.; Fargnoli, J. Abstract B192: Preclinical characterization of BMS-833923 (XL139), a hedgehog (HH) pathway inhibitor in early clinical development. Mol. Cancer Ther. 2009, 8. [Google Scholar] [CrossRef]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2011, 3, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Trang, V.; Lee, A.; Williams, N.S.; Wilson, A.N.; Epstein, E.H., Jr.; Tang, J.Y.; Kim, J. Posaconazole, a Second-Generation Triazole Antifungal Drug, Inhibits the Hedgehog Signaling Pathway and Progression of Basal Cell Carcinoma. Mol. Cancer Ther. 2016, 15, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, T.; Oguro, Y.; Tanaka, T.; Shiokawa, Z.; Tanaka, Y.; Shibata, S.; Sato, Y.; Yamakawa, H.; Hattori, H.; Yamamoto, Y.; et al. Discovery of the investigational drug TAK-441, a pyrrolo[3,2-c]pyridine derivative, as a highly potent and orally active hedgehog signaling inhibitor: Modification of the core skeleton for improved solubility. Bioorg. Med. Chem. 2012, 20, 5507–5517. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Guicherit, O.M.; Zaharian, B.I.; Xu, Y.; Chai, L.; Wichterle, H.; Kon, C.; Gatchalian, C.; Porter, J.A.; Rubin, L.L.; et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: Effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 4616–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohner, A.; Spilker, M.E.; Lam, J.L.; Pascual, B.; Bartkowski, D.; Li, Q.J.; Yang, A.H.; Stevens, G.; Xu, M.; Wells, P.A.; et al. Effective targeting of Hedgehog signaling in a medulloblastoma model with PF-5274857, a potent and selective Smoothened antagonist that penetrates the blood-brain barrier. Mol. Cancer Ther. 2012, 11, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Jin, Q.; Koo, D.I.; Liao, X.; Englund, N.P.; Wang, Y.; Ramamurthy, A.; Schultz, P.G.; Dorsch, M.; Kelleher, J.; et al. Small molecule antagonists in distinct binding modes inhibit drug-resistant mutant of smoothened. Chem. Biol. 2011, 18, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chaudhary, A.K.; Dong, Y.; Zhong, H.A.; Mondal, G.; Lin, F.; Kumar, V.; Mahato, R.I. Design, Synthesis and Biological Evaluation of novel Hedgehog Inhibitors for treating Pancreatic Cancer. Sci. Rep. 2017, 7, 1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roudaut, H.; Traiffort, E.; Gorojankina, T.; Vincent, L.; Faure, H.; Schoenfelder, A.; Mann, A.; Manetti, F.; Solinas, A.; Taddei, M.; et al. Identification and mechanism of action of the acylguanidine MRT-83, a novel potent Smoothened antagonist. Mol. Pharmacol. 2011, 79, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Hoch, L.; Faure, H.; Roudaut, H.; Schoenfelder, A.; Mann, A.; Girard, N.; Bihannic, L.; Ayrault, O.; Petricci, E.; Taddei, M.; et al. MRT-92 inhibits Hedgehog signaling by blocking overlapping binding sites in the transmembrane domain of the Smoothened receptor. FASEB J. 2015, 29, 1817–1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrobono, S.; Santini, R.; Gagliardi, S.; Dapporto, F.; Colecchia, D.; Chiariello, M.; Leone, C.; Valoti, M.; Manetti, F.; Petricci, E.; et al. Targeted inhibition of Hedgehog-GLI signaling by novel acylguanidine derivatives inhibits melanoma cell growth by inducing replication stress and mitotic catastrophe. Cell Death Dis. 2018, 9, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, V.M.; Chen, S.C.; Arkin, M.R.; Reiter, J.F. Small molecule inhibitors of Smoothened ciliary localization and ciliogenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 13644–13649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Davidow, L.; Arvanites, A.C.; Blanchard, J.; Lam, K.; Xu, K.; Oza, V.; Yoo, J.W.; Ng, J.M.; Curran, T.; et al. Glucocorticoid compounds modify smoothened localization and hedgehog pathway activity. Chem. Biol. 2012, 19, 972–982. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Arvanites, A.C.; Davidow, L.; Blanchard, J.; Lam, K.; Yoo, J.W.; Coy, S.; Rubin, L.L.; McMahon, A.P. Selective identification of hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation. ACS Chem. Biol. 2012, 7, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Kremer, L.; Schultz-Fademrecht, C.; Baumann, M.; Habenberger, P.; Choidas, A.; Klebl, B.; Kordes, S.; Schöler, H.R.; Sterneckert, J.; Ziegler, S.; et al. Discovery of a Novel Inhibitor of the Hedgehog Signaling Pathway through Cell-based Compound Discovery and Target Prediction. Angew. Chem. Int. Ed. Engl. 2017, 56, 13021–13025. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; He, Y.; Chen, G.; Ma, H.; Zheng, J.; Zhang, Z.; Cao, B.; Zhang, H.; Zhang, X.; Mao, X. A novel hedgehog inhibitor for the treatment of hematological malignancies. Anticancer Drugs 2018, 29, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Mook, R.A., Jr.; Lu, J.; Gooden, D.M.; Ribeiro, A.; Guo, A.; Barak, L.S.; Lyerly, H.K.; Chen, W. Identification of a novel Smoothened antagonist that potently suppresses Hedgehog signaling. Bioorg. Med. Chem. 2012, 20, 6751–6757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferruzzi, P.; Mennillo, F.; De Rosa, A.; Giordano, C.; Rossi, M.; Benedetti, G.; Magrini, R.; Pericot Mohr, G.L.; Miragliotta, V.; Magnoni, L.; et al. In vitro and in vivo characterization of a novel Hedgehog signaling antagonist in human glioblastoma cell lines. Int. J. Cancer 2012, 131, E33–E44. [Google Scholar] [CrossRef] [PubMed]
- Dockendorff, C.; Nagiec, M.M.; Weïwer, M.; Buhrlage, S.; Ting, A.; Nag, P.P.; Germain, A.; Kim, H.J.; Youngsaye, W.; Scherer, C.; et al. Macrocyclic Hedgehog Pathway Inhibitors: Optimization of Cellular Activity and Mode of Action Studies. ACS Med. Chem. Lett. 2012, 3, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.M.; Wang, D.C.; Xiang, P.; Zhang, J.N.; Sang, Y.X.; Lin, H.J.; Chen, J.; Xie, G.; Song, H.; Zhao, Y.L.; et al. Synthesis and biological evaluation of novel benzamide derivatives as potent smoothened antagonists. Bioorg. Med. Chem. Lett. 2014, 24, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Chen, K.; Song, B.; Deng, C.; Li, W.; Niu, L.; Bai, M.; Song, H.; Zhang, L. Synthesis and Smo Activity of Some Novel Benzamide Derivatives. Molecules 2017, 23, 85. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Liu, Y.; Ma, H.; Zheng, J.; Tian, S.; Sun, Z.; Luo, L.; Li, J.; Zhang, H.; Yang, Z.J.; et al. Design, Synthesis, and Structure-Activity Relationship of Tetrahydropyrido[4,3-d]pyrimidine Derivatives as Potent Smoothened Antagonists with in Vivo Activity. ACS Chem. Neurosci. 2017, 8, 1980–1994. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Lu, W.; Sun, Z.; Luo, L.; Geng, D.; Yang, Z.; Li, E.; Zheng, J.; Wang, M.; Zhang, H.; et al. Design, synthesis, and structure–activity-relationship of tetrahydrothiazolopyridine derivatives as potent smoothened antagonists. Eur. J. Med. Chem. 2015, 89, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, D.; Jung, J.H.; Han, S.; Lee, H.; Oh, S.J.; Ko, H.W.; Lee, K. Design, synthesis, and biological evaluation of structurally modified isoindolinone and quinazolinone derivatives as hedgehog pathway inhibitors. Eur. J. Med. Chem. 2017, 125, 1036–1050. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Han, D.; Gao, W.; Jing, Q.; Jiang, J.; Wan, Y.; Englund, N.P.; Tuntland, T.; Wu, X.; Pan, S. Design, synthesis, and structure-activity-relationship of phenyl imidazoles as potent Smoothened antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 6573–6576. [Google Scholar] [CrossRef] [PubMed]
- Muraglia, E.; Ontoria, J.M.; Branca, D.; Dessole, G.; Bresciani, A.; Fonsi, M.; Giuliano, C.; Llauger Bufi, L.; Monteagudo, E.; Palumbi, M.C.; et al. N-(2-alkylaminoethyl)-4-(1,2,4-oxadiazol-5-yl)piperazine-1-carboxamides as highly potent smoothened antagonists. Bioorg. Med. Chem. Lett. 2011, 21, 5283–5288. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Bufi, L.L.; Torrisi, C.; Bresciani, A.; Giomini, C.; Rowley, M.; Serafini, S.; Bin, H.; Hao, W.; Steinkühler, C.; et al. Identification of a series of 4-[3-(quinolin-2-yl)-1,2,4-oxadiazol-5-yl]piperazinyl ureas as potent smoothened antagonist hedgehog pathway inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5274–5282. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Xue, D.; Yang, J.; Wang, J.; Liu, X.; Huang, W.; Li, J.; Long, Y.Q.; Tan, W.; Zhang, A. Design, Synthesis, and Pharmacological Evaluation of 2-(2,5-Dimethyl-5,6,7,8-tetrahydroquinolin-8-yl)-N-aryl Propanamides as Novel Smoothened (Smo) Antagonists. J. Med. Chem. 2016, 59, 11050–11068. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.R.; Zhao, H.; Zhang, X.S.; Lang, H.; Yu, K. Novel-smoothened inhibitors for therapeutic targeting of naïve and drug-resistant hedgehog pathway-driven cancers. Acta Pharmacol. Sin. 2018. [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Toftgård, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoya, T.; Arai, M.A.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. Naturally occurring small-molecule inhibitors of hedgehog/GLI-mediated transcription. Chembiochem 2008, 9, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.C.; Peaceman, D.; Ozdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Nanta, R.; Kumar, D.; Meeker, D.; Rodova, M.; Van Veldhuizen, P.J.; Shankar, S.; Srivastava, R.K. NVP-LDE-225 (Erismodegib) inhibits epithelial-mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rγ null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2013, 2, e42. [Google Scholar] [CrossRef] [PubMed]
- Jalili, A.; Mertz, K.D.; Romanov, J.; Wagner, C.; Kalthoff, F.; Stuetz, A.; Pathria, G.; Gschaider, M.; Stingl, G.; Wagner, S.N. NVP-LDE225, a potent and selective SMOOTHENED antagonist reduces melanoma growth in vitro and in vivo. PLoS ONE 2013, 8, e69064. [Google Scholar] [CrossRef]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dummer, R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kaatz, M.; Loquai, C.; Stratigos, A.J.; et al. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2016, 75, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Swiss Group for Clinical Cancer Research (SAKK). Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.T.; Nadesan, P.; Ali, S.A.; Wang, C.Y.; Whetstone, H.; Poon, R.; Wei, Q.; Keilty, J.; Proctor, J.; Wang, L.W.; et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor IPI-926 directly inhibits sarcoma cell growth. Mol. Cancer Ther. 2014, 13, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Hoyt, J.; Whitebread, N.; Manna, J.; Peluso, M.; Faia, K.; Campbell, V.; Tremblay, M.; Nair, S.; Grogan, M.; et al. The pre-clinical absorption, distribution, metabolism and excretion properties of IPI-926, an orally bioavailable antagonist of the hedgehog signal transduction pathway. Xenobiotica 2013, 43, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Discontinued drugs in 2012: Oncology drugs. Expert Opin. Investig. Drugs 2013, 22, 1627–1644. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, A.H.; Komatsu, Y.; Kelly, L.A.; Malhotra, U.; Rotoloni, C.; Kosovec, J.E.; Zahoor, H.; Makielski, R.; Bhatt, A.; Hoppo, T.; et al. Smoothened inhibition leads to decreased proliferation and induces apoptosis in esophageal adenocarcinoma cells. Cancer Investig. 2013, 31, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmöller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Li, S.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, G.; Oehler, V.G.; Papayannidis, C.; Courtney, R.; Shaik, M.N.; Zhang, X.; O’Connell, A.; McLachlan, K.R.; Zheng, X.; Radich, J.; et al. Treatment with PF-04449913, an oral smoothened antagonist, in patients with myeloid malignancies: A phase 1 safety and pharmacokinetics study. Lancet Haematol. 2015, 2, e339–e346. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naïve and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. TAK-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013, 133, 1955–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Shimizu, Y.; Nakashima, K.; Kondo, S.; Ogawa, K.; Sasaki, S.; Matsui, H. Inhibition mechanism exploration of investigational drug TAK-441 as inhibitor against Vismodegib-resistant Smoothened mutant. Eur. J. Pharmacol. 2014, 723, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Williams, R. Discontinued in 2013: Oncology drugs. Expert Opin. Investig. Drugs 2015, 24, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Xiao, T.Z.; Oda, Y.; Chang, K.S.; Shpall, E.; Wu, A.; So, P.L.; Hebert, J.; Bikle, D.; Epstein, E.H., Jr. Vitamin D3 inhibits hedgehog signaling and proliferation in murine Basal cell carcinomas. Cancer Prev. Res. 2011, 4, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Brinkhuizen, T.; Frencken, K.J.; Nelemans, P.J.; Hoff, M.L.; Kelleners-Smeets, N.W.; Zur Hausen, A.; van der Horst, M.P.; Rennspiess, D.; Winnepenninckx, V.J.; van Steensel, M.A.; et al. The effect of topical diclofenac 3% and calcitriol 3 μg/g on superficial basal cell carcinoma (sBCC) and nodular basal cell carcinoma (nBCC): A phase II, randomized controlled trial. J. Am. Acad. Dermatol. 2016, 75, 126–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romer, J.T.; Kimura, H.; Magdaleno, S.; Sasai, K.; Fuller, C.; Baines, H.; Connelly, M.; Stewart, C.F.; Gould, S.; Rubin, L.L.; et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/-)p53(-/-) mice. Cancer Cell 2004, 6, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Guerlet, G.; Spangenberg, T.; Mann, A.; Faure, H.; Ruat, M. Synthesis and biological evaluation of desmethylveramiline; a micromolar Hedgehog inhibitor. Bioorg. Med. Chem. Lett. 2011, 21, 3608–3612. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Aftab, B.T.; Tang, J.Y.; Kim, D.; Lee, A.H.; Rezaee, M.; Kim, J.; Chen, B.; King, E.M.; Borodovsky, A.; et al. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell 2013, 23, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Manetti, F.; Faure, H.; Roudaut, H.; Gorojankina, T.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Solinas, A.; Taddei, M.; Ruat, M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as smoothened antagonists. Mol. Pharmacol. 2010, 78, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Solinas, A.; Faure, H.; Roudaut, H.; Traiffort, E.; Schoenfelder, A.; Mann, A.; Manetti, F.; Taddei, M.; Ruat, M. Acylthiourea, acylurea, and acylguanidine derivatives with potent hedgehog inhibiting activity. J. Med. Chem. 2012, 55, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Chiarenza, A.; Manetti, F.; Petricci, E.; Ruat, M.; Naldini, A.; Taddei, M.; Carraro, F. Novel Acylguanidine Derivatives Targeting Smoothened Induce Antiproliferative and Pro-Apoptotic Effects in Chronic Myeloid Leukemia Cells. PLoS ONE 2016, 11, e0149919. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, G.; Geminiani, M.; Gambassi, S.; Orlandini, M.; Petricci, E.; Marzocchi, B.; Laschi, M.; Taddei, M.; Manetti, F.; Santucci, A. Novel smoothened antagonists as anti-neoplastic agents for the treatment of osteosarcoma. J. Cell. Physiol. 2018, 233, 4961–4971. [Google Scholar] [CrossRef] [PubMed]
- Vesci, L.; Milazzo, F.M.; Stasi, M.A.; Pace, S.; Manera, F.; Tallarico, C.; Cini, E.; Petricci, E.; Manetti, F.; De Santis, R.; et al. Hedgehog pathway inhibitors of the acylthiourea and acylguanidine class show antitumor activity on colon cancer in vitro and in vivo. Eur. J. Med. Chem. 2018, 157, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Brinkhuizen, T.; Reinders, M.G.; van Geel, M.; Hendriksen, A.J.; Paulussen, A.D.; Winnepenninckx, V.J.; Keymeulen, K.B.; Soetekouw, P.M.; van Steensel, M.A.; Mosterd, K. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2014, 71, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Ponomaryov, T.; Ornell, K.J.; Zhou, P.; Dabral, S.K.; Pak, E.; Li, W.; Atwood, S.X.; Whitson, R.J.; Chang, A.L.; et al. RAS/MAPK Activation Drives Resistance to Smo Inhibition, Metastasis, and Tumor Evolution in Shh Pathway-Dependent Tumors. Cancer Res. 2015, 75, 3623–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, K.E.; de Miera, E.V.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals 2013, 6, 1429–1450. [Google Scholar] [CrossRef] [PubMed]
- Pogoriler, J.; Millen, K.; Utset, M.; Du, W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 2006, 133, 3929–3937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCubrey, J.A.; Steelman, L.S.; Bertrand, F.E.; Davis, N.M.; Sokolosky, M.; Abrams, S.L.; Montalto, G.; D’Assoro, A.B.; Libra, M.; Nicoletti, F.; et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget 2014, 5, 2881–2911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellino, R.C.; Barwick, B.G.; Schniederjan, M.; Buss, M.C.; Becher, O.; Hambardzumyan, D.; Macdonald, T.J.; Brat, D.J.; Durden, D.L. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS ONE 2010, 5, e10849. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Ma, H.; Sun, Z.; Luo, L.; Tian, S.; Zheng, J.; Zhang, X. Discovery of a 6-(pyridin-3-yl)benzo[d]thiazole template for optimization of hedgehog and PI3K/AKT/mTOR dual inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 3665–3670. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Gruber, W.; Hutzinger, M.; Elmer, D.P.; Parigger, T.; Sternberg, C.; Cegielkowski, L.; Zaja, M.; Leban, J.; Michel, S.; Hamm, S.; et al. DYRK1B as therapeutic target in Hedgehog/GLI-dependent cancer cells with Smoothened inhibitor resistance. Oncotarget 2016, 7, 7134–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Coni, S.; Mancuso, A.B.; Di Magno, L.; Sdruscia, G.; Manni, S.; Serrao, S.M.; Rotili, D.; Spiombi, E.; Bufalieri, F.; Petroni, M.; et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH Medulloblastoma. Sci. Rep. 2017, 7, 44079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Quan, H.; Xie, C.; Lou, L. NL-103, a novel dual-targeted inhibitor of histone deacetylases and hedgehog pathway, effectively overcomes vismodegib resistance conferred by Smo mutations. Pharmacol. Res. Perspect. 2014, 2, e00043. [Google Scholar] [CrossRef] [PubMed]
- Chun, S.G.; Park, H.; Pandita, R.K.; Horikoshi, N.; Pandita, T.K.; Schwartz, D.L.; Yordy, J.S. Targeted inhibition of histone deacetylases and hedgehog signaling suppress tumor growth and homologous recombination in aerodigestive cancers. Am. J. Cancer Res. 2015, 5, 1337–1352. [Google Scholar] [PubMed]
- Mujtaba, S.; Zeng, L.; Zhou, M.M. Structure and acetyl-lysine recognition of the bromodomain. Oncogene 2007, 26, 5521–5527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.H.; Ayad, N.G.; et al. The BET bromodomain inhibitor I-BET151 acts downstream of smoothened protein to abrogate the growth of hedgehog protein-driven cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef] [PubMed]
- Whitson, R.J.; Lee, A.; Urman, N.M.; Mirza, A.; Yao, C.Y.; Brown, A.S.; Li, J.R.; Shankar, G.; Fry, M.A.; Atwood, S.X.; et al. Noncanonical hedgehog pathway activation through SRF-MKL1 promotes drug resistance in basal cell carcinomas. Nat. Med. 2018, 24, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates GLI1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Vanner, R.J.; Remke, M.; Gallo, M.; Selvadurai, H.J.; Coutinho, F.; Lee, L.; Kushida, M.; Head, R.; Morrissy, S.; Zhu, X.; et al. Quiescent sox2(+) cells drive hierarchical growth and relapse in sonic hedgehog subgroup medulloblastoma. Cancer Cell 2014, 26, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Walsh, M.P.; Ali, S.A.; Thompson, E.A.; Murray, N.R.; Fields, A.P. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014, 25, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; de Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Danés, A.; Larsimont, J.C.; Liagre, M.; Muñoz-Couselo, E.; Lapouge, G.; Brisebarre, A.; Dubois, C.; Suppa, M.; Sukumaran, V.; Del Marmol, V.; et al. A slow-cycling LGR5 tumour population mediates basal cell carcinoma relapse after therapy. Nature 2018, 562, 434–438. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Role | Cancer Type | Reference |
|---|---|---|---|
| S278I | oncogenic | BCC, MB | [78,79] |
| L412F | oncogenic | BCC, Ameloblastoma, Meningioma | [78,80,81,82] |
| S533N | oncogenic | PNET | [83] |
| W535L (SMO-M2) | oncogenic | BCC, Ameloblastoma, Meningioma | [14,81,82] |
| R562Q (SMO-M1) | oncogenic | BCC | [14] |
| N219D (mN223D) | sonidegib-resistance | Ptch+/−; p53−/− MB | [84] |
| L221R (mL225R) | sonidegib-resistance | Ptch+/−; p53−/− MB | [84] |
| D384N (mD388N) | sonidegib-resistance | Ptch+/−; p53−/− MB | [84] |
| S387N (mS391N) | sonidegib-resistance | Ptch+/−; p53−/− MB | [84] |
| G453S (mG457S) | sonidegib-resistance | Ptch+/−; p53−/− MB | [84] |
| H231R | vismodegib-resistance | BCC | [78] |
| T241M | vismodegib-resistance | BCC | [80] |
| W281C | vismodegib-resistance | BCC | [78,80] |
| V321M | vismodegib-resistance | BCC | [78,80] |
| I408V | vismodegib-resistance | BCC | [80] |
| L412F | vismodegib-resistance | BCC | [78,80] |
| A459V | vismodegib-resistance | BCC | [80] |
| F460L | vismodegib-resistance | BCC | [78] |
| C469Y | vismodegib-resistance | BCC | [80] |
| D473H* | vismodegib-resistance | MB | [65] |
| D473G* | vismodegib-resistance | BCC | [78,85] |
| D473Y | vismodegib-resistance | BCC | [86] |
| Q477E* | vismodegib-resistance | BCC | [78] |
| G497W | vismodegib primary res. | BCC | [86] |
| E518K/A | vismodegib-resistance | [66] | |
| S533N | vismodegib-resistance | BCC | [80] |
| W535L* | vismodegib-resistance | BCC | [78,80,85] |
| Q635E | vismodegib-resistance | BCC | [78] |
| Pathway Antagonists | Mechanism of Action | Status | Reference |
|---|---|---|---|
| At SMO Level | |||
| Cyclopamine | Binds 7TM domain | Preclinical | [3] |
| KAAD-Cyclopamine | Binds 7TM domain | Preclinical | [3] |
| IPI-269609 | Binds 7TM domain | Preclinical | [92] |
| GDC-0449 (Vismodegib) | Binds 7TM domain | 68 Clinical trials | [98] |
| LDE-225 (Sonidegib) | Binds 7TM domain | 37 Clinical trials | [99] |
| IPI-926 (Saridegib) | Binds 7TM domain | 6 Clinical trials | [100] |
| BMS-833923/XL139 | Binds 7TM domain | 8 Clinical trials | [101] |
| PF-04449913 (Glasdegib) | Binds 7TM domain | 11 Clinical trials | [102] |
| LY2940680 (Taladegib) | Binds 7TM domain | 6 Clinical trials | [68] |
| Itraconazole | Binds SMO (BS distinct from Cyc) | 48 Clinical trials | [103] |
| Posaconazole | Binds SMO (BS distinct from Cyc) | 16 Clinical trials | [104] |
| TAK-441 | 1 Clinical trial | [105] | |
| LEQ-506 | 1 Clinical trial | [67] | |
| Vitamin D3 | Binds 7TM domain | 3 Clinical trials | [73] |
| Cur-61414 | Preclinical | [106] | |
| PF-5274857 | Preclinical | [107] | |
| Compound 5 | Preclinical | [66] | |
| SANT1-4 | Bind 7TM domain | Preclinical | [60] |
| ALLO 1-2 | Bind extracellular CRD | Preclinical | [108] |
| DMB5 | Binds 7TM domain | Preclinical | [109] |
| MRT-83 | Binds 7TM domain | Preclinical | [110] |
| MRT-92 | Binds 7TM domain | Preclinical | [111,112] |
| SA1-10 | Inhibit SMO ciliary localization | Preclinical | [113] |
| Budesonide | Inhibits SMO ciliary translocation | Preclinical | [114] |
| SMANT | Inhibits SMO ciliary translocation | Preclinical | [115] |
| DY131 | Inhibits SMO ciliary translocation | Preclinical | [115] |
| Smoothib | Preclinical | [116] | |
| HH78 | Preclinical | [117] | |
| A8 | Preclinical | [118] | |
| SEN450 | Preclinical | [119] | |
| BRD-6851 | Preclinical | [120] | |
| Benzamide derivatives | Preclinical | [121,122] | |
| Tetrahydropyridopyrimidine derivatives | Preclinical | [123] | |
| Tetrahydrothiazolopyridine derivatives | Preclinical | [124] | |
| Quinazolinone derivatives | Preclinical | [125] | |
| Phenyl imidazole derivatives | Preclinical | [126] | |
| Piperazine-1-carboxamides | Preclinical | [127] | |
| Piperazinyl urea derivatives | [128] | ||
| N-arylpropanamide | Preclinical | [129] | |
| Benzimidazole derivatives | Preclinical | [130] | |
| Downstream of SMO | |||
| GANT58-61 | Inhibit GLI-mediated luciferase | Preclinical | [131] |
| Arcyriaflavin C | Inhibits GLI-mediated luciferase | Preclinical | [132] |
| Physalin F | Inhibits GLI-mediated luciferase | Preclinical | [132] |
| HPI1-4 | Modulate GLI activation | Preclinical | [133] |
| ATO | Inhibits GLI transcription factors | 41 Clinical trials | [134,135] |
| Pyrvinium | Enhances GLI degradation | Preclinical | [136] |
| Glabrescione B | Interferes with DNA binding | Preclinical | [137] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietrobono, S.; Stecca, B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells 2018, 7, 272. https://doi.org/10.3390/cells7120272
Pietrobono S, Stecca B. Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells. 2018; 7(12):272. https://doi.org/10.3390/cells7120272
Chicago/Turabian StylePietrobono, Silvia, and Barbara Stecca. 2018. "Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors" Cells 7, no. 12: 272. https://doi.org/10.3390/cells7120272
APA StylePietrobono, S., & Stecca, B. (2018). Targeting the Oncoprotein Smoothened by Small Molecules: Focus on Novel Acylguanidine Derivatives as Potent Smoothened Inhibitors. Cells, 7(12), 272. https://doi.org/10.3390/cells7120272