Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence



{kind=link}
Abstract
:1. Function of Lamin B Receptor in Anchoring Chromatin to the Inner Nuclear Membrane
2. Distinct Types of Heterochromatin Attachment to the Nuclear Membrane in Undifferentiated (Embryonic) and Differentiated Cells
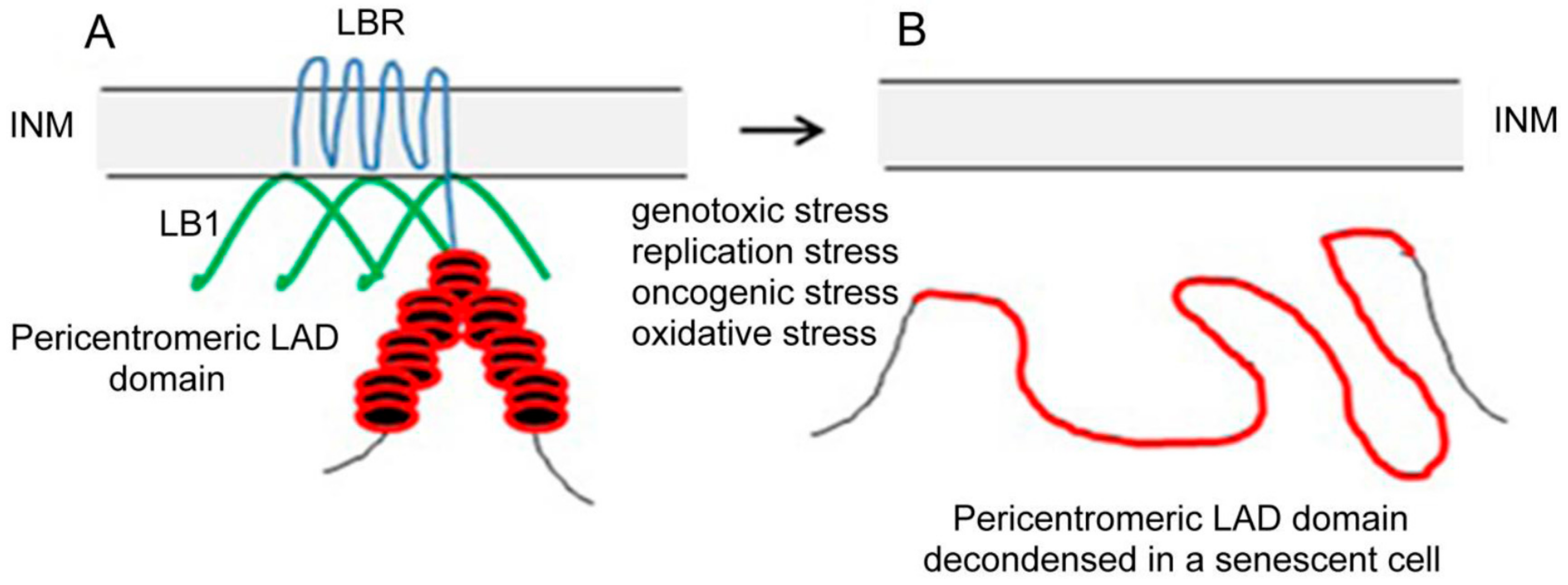
3. Lamin B is Downregulated in Senescence
4. The Onset of Senescence is Accompanied by a Coordinated Downregulation of Lamin B Receptor and Lamin B1 Expression
5. Consequences of LBR Downregulation in Senescence
6. Conclusions
Acknowledgements
Conflicts of Interest
References
- Woodcock, C.L.; Ghosh, R.P. Chromatin higher order structure and dynamics. Cold Spring Harb. Perspect. Biol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Guelen, L.; Pagie, L.; Brasset, E.; Meuleman, W.; Faza, M.B.; Talhout, W.; Eussen, B.H.; de Klein, A.; Wessels, L.; de Laat, W.; et al. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions in single human cells. Nature 2008, 453, 948–951. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Wang, A.S.; Thanisch, K.; Schmidt, C.S.; Krebs, S.; Zwerger, M.; Cohen, T.V.; Devys, D.; Foisner, R.; Peichl, L.; et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 2013, 152, 584–598. [Google Scholar] [CrossRef] [PubMed]
- Andrés, V.; González, J.M. Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 2009, 187, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Krohne, G. LEM-Domain proteins: New insights into lamin-interacting proteins. Int. Rev. Cytol. 2007, 216, 1–46. [Google Scholar]
- Brachner, A.; Foisner, R. Evolvement of LEM proteins as chromatin tethers at the nuclear periphery. Biochem. Soc. Trans. 2011, 39, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Makatsori, D.; Kourmouli, N.; Polioudaki, H.; Shultz, L.D.; McLean, K.; Theodoropoulos, P.A.; Singh, P.B.; Georgatos, S.D. The inner nuclear membrane protein lamin B receptor forms distinct microdomains and links epigenetically marked chromatin to the nuclear envelope. J. Biol. Chem. 2004, 279, 25567–25573. [Google Scholar] [CrossRef] [PubMed]
- Olins, A.L.; Rhodes, G.; Welch, D.B.; Zwerger, M.; Olins, D.E. Lamin B receptor: Multi-tasking at the nuclear envelope. Nucleus 2010, 1, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Hirano, Y.; Hizume, K.; Kimura, H.; Takeyasu, K.; Haraguchi, T.; Hiraoka, Y. Lamin B receptor recognizes specific modifications of histone H4 in heterochromatin formation. J. Biol. Chem. 2012, 287, 42654–42663. [Google Scholar] [CrossRef] [PubMed]
- Clowney, E.J.; LeGros, M.A.; Mosley, C.P.; Clowney, F.G.; Markenskoff-Papadimitriou, E.C.; Myllys, M.; Barnea, G.; Larabell, C.A.; Lomvardas, S. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell 2012, 151, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Sharov, A.A.; McDole, K.; Cheng, M.; Hao, A.; Fan, C.M.; Giano, M.N.; Ko, M.S.; Zheng, Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science 2011, 334, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Chang, S.Y.; Yin, L.; Tu, Y.; Hu, Y.; Yoshinaga, Y.; de Jong, P.J.; Fong, L.G.; Young, S.G. An absence of both lamin B1 and lamin B2 in keratinocytes has no effect on cell proliferation or the development of skin and hair. Hum. Mol. Genet. 2011, 20, 3537–3544. [Google Scholar] [CrossRef] [PubMed]
- Broers, J.L.; Ramaekers, F.C.; Bonne, G.; Yaou, R.B.; Hutchison, C.J. Nuclear lamins: Laminopathies and their role in premature ageing. Phisiol. Rev. 2006, 86, 967–1008. [Google Scholar] [CrossRef] [PubMed]
- Holmer, L.; Worman, H.J. Inner nuclear membrane proteins: Functions and targeting. Cell. Mol. Life Sci. 2001, 58, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, K.; Egelhofer, T.A.; Strome, S.; Lieb, J.D. Caenorhabditis chromosome arms are anchored to nuclear membrane via discontinuous association with LEM-2. Genome Biol. 2010, 11, R120. [Google Scholar] [CrossRef] [PubMed]
- Mattout, A.; Pike, B.L.; Towbin, B.D.; Bank, E.M.; Gonzales-Sandoval, A.; Stadier, M.B.; Meister, P.; Gruenbaum, Y.; Gasser, S.M. An EDMD mutation in C. elegans lamin blocks muscle-specific gene relocation and compromises muscle integrity. Curr. Biol. 2011, 21, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Towbin, B.D.; Gonzáles-Aguilera, C.; Sack, R.; Gaidatzis, D.; Kalck, V.; Meister, P.; Askjaer, P.; Gasser, S.M. Step-wise methylation of histone H3K9 positions heterochromatin at the nuclear periphery. Cell 2012, 150, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Novakova, Z.; Hubackova, S.; Kosar, M.; Janderova-Rossmeislova, L.; Dobrovolna, J.; Vasicova, P.; Vancurova, M.; Horejsi, Z.; Hozak, P.; Bartek, J.; et al. Cytokine expression and signaling in drug-induced cellular senescence. Oncogene 2010, 29, 273–284. [Google Scholar] [CrossRef] [PubMed]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T.; Saretzki, G.; Ladhoff, J.; d’Adda di Fagagna, F.; Jackson, S.P. Human cell senescence as a DNA damage response. Mech. Ageing Dev. 2005, 126, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M. Telomeres limit cancer growth by inducing senescence: Long-sought in vivo evidence obtained. Cancer Cell 2007, 11, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Kosar, M.; Bartkova, J.; Hubackova, S.; Hodny, Z.; Lukas, J.; Bartek, J. Senescence-asociated heterochromatin foci are dispensable for cellular senescence, occur in a cell type- and insult-dependent manner and follow expression of p16ink4a. Cell Cycle 2011, 10, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.M.; Bartholomew, J.C.; Campisi, J.; Acosta, M.; Reagan, J.D.; Ames, B.N. Molecular analysis of H2O2-induced senescent-like growth arrest in normal human fibroblasts: P53 and Rb control G1 arrest but not cellreplication. Biochem. J. 1998, 332, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Lintos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Lukášová, E.; Kovařík, A.; Bačíková, A.; Falk, M.; Kozubek, S. Loss of lamin B receptor is necessary to induce cellular senescence. Biochem. J. 2017, 474, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromati foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed]
- Shimi, T.; Butin-Israeli, V.; Adam, S.A.; Hamanaka, R.B.; Goldman, A.E.; Lucas, C.A.; Shumaker, D.K.; Kosak, S.T.; Chandel, N.S.; Goldman, R.D. The role of lamin B1 in cell proliferation and senescence. Genes Dev. 2011, 25, 2579–2593. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Chojnowski, A.; Ong, P.F.; Zhao, T.Y.; Common, J.E.; Lunny, D.; Lane, E.B.; Lee, S.J.; Vardy, L.A.; Stewart, C.L.; et al. Lamin B1 fluctuations have differential effects on cellular proliferation and senescence. J. Cell Biol. 2013, 200, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescence cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.-Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescence cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Chandra, T.; Kirschner, K.; Thuret, J.-Y.; Pope, B.J.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of repressive histone markers and chromatin compaction during senescent heterochromatic layer formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Sadaie, M.; Salama, R.; Carroll, T.; Tomimatsu, K.; Chandra, T.; Young, A.R.J.; Narita, M.; Pérez-Mancera, P.A.; Bennett, D.C.; Chong, H.; et al. Redistribution of the lamin B1 genomic binding profile affects rearrangement of heterochromatic domains and SAHF formation during senescence. Genes Dev. 2015, 27, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Worman, H.J. Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane. J. Biol. Chem. 1994, 269, 11306–11311. [Google Scholar] [PubMed]
- Worman, H.J.; Yuan, J.; Blobel, G.; Georgatos, S.P. A lamin B receptor in the nuclear envelope. Proc. Natl. Acad. Sci. USA 1988, 85, 8531–8534. [Google Scholar] [CrossRef] [PubMed]
- Ye, Q.; Worman, H.J. Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins to Drosophilla HP1. J. Biol. Chem. 1996, 271, 14653–14656. [Google Scholar] [CrossRef] [PubMed]
- Von Mikecz, A.; Chen, M.; Rockel, T.; Scharf, A. The nuclear ubiquitin-proteasome system: Visualization of proteasomes, protein aggregates, and proteolysis in the cell nucleus. Nucleus 2008, 463, 191–202. [Google Scholar]
- De Cecco, M.; Criscione, S.W.; Peckham, E.J.; Hillenmeyer, S.; Hamm, E.A.; Manivannan, J.; Peterson, A.L.; Kreiling, J.A.; Neretti, N.; Sedivy, J.M. Genomes of replicative senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell 2013, 12, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Malhas, A.; Lee, C.F.; Sanders, R.; Sounders, N.J.; Vaux, D.J. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J. Cell Biol. 2007, 176, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Nikolakaki, E.; Meier, J.; Simons, G.; Georgatos, S.D.; Giannakouros, T. Mitotic phosphorylation of the lamin B receptor by a serine/arginine kinase and p34(cdc2). J. Biol. Chem. 1997, 272, 6208–6213. [Google Scholar] [CrossRef]
- Duband-Goulet, I.; Courvalin, J.-C.; Buendia, B. LBR, a chromatin and lamin binding protein from the inner nuclear membrane, is proteolysed at late stage of apoptosis. J. Cell Sci. 1998, 111, 1441–1451. [Google Scholar] [PubMed]
- Ellenberg, J.; Siggia, E.D.; Moreira, J.E.; Smith, C.L.; Presley, J.F.; Worman, H.J.; Lippincott-Schwartz, J. Nuclear membrane dynamics and reassembly in living cells: Targeting of an inner nuclear membrane protein in interphase and mitosis. J. Cell Biol. 1997, 138, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Nikolakaki, E.; Milonis, I.; Giannakouros, T. Lamin B receptor: Interplay between structure, function and localization. Cells 2017, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Tseng, L.C.; Chen, R.H. Temporal control of nuclear envelope assembly by phosphorylation of lamin B receptor. Mol. Biol. Cell 2011, 22, 3306–3317. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.S.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysozome-mediated processing of chromatin in senescence. J. Cell Biol. 2012, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Peric-Hupkes, D.; Meuleman, W.; Pagie, L.; Bruggeman, S.W.; Solovei, I.; Brugman, W.; Graf, S.; Flicek, P.; Kerkhoven, R.M.; Reinders, M.; et al. Molecular maps of the reorganization of genom-nuclear lamina interactions during differentiation. Mol. Cell 2010, 38, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Meulman, W.; Peric-Hupkes, D.; Kind, J.; Beaudry, J.B.; Pagie, L.; Kellis, M.; Reinders, M.; Wessels, L.; van Steensel, B. Constitutive nuclear lamina-genomeinteractions are highly conserved and associated with A/T-rich sequence. Genome Res. 2013, 23, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Thanisch, K.; Feodorova, Y. How to rule the nucleus: Divide et impera. Curr. Opin. Cell Biol. 2016, 40, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Geeven, G.; Eyres, M.; Bergsma, A.J.; Janssen, M.; Versteegen, M.; Zhu, Y.; Schell, Y.; Vermeulen, C.; de Vit, E.; et al. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015, 25, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Schemelleh, L.; During, K.; Engelhardt, A.; Stein, S.; Cremer, C.; Cremer, T. Differences in centromere positioning of cycling and postmitotic human cell types. Chromosoma 2004, 112, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, B.A.; de Laat, W. Getting the genome in shape: The formation of loops, domains and compartments. Genome Biol. 2015, 16, 154. [Google Scholar] [CrossRef] [PubMed]
- Weierich, C.; Brero, A.; Stein, S.; von Hase, J.; Cremer, C.; Cremer, T.; Solovei, I. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosom. Res. 2003, 11, 485–502. [Google Scholar] [CrossRef]
- Swanson, E.C.; Manning, B.; Zhang, H.; Lawrence, J.B. Higher-order unfolding of satellite heterochromatin is a consistent and early event in cell senescence. J. Cell Biol. 2013, 203, 929–942. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lukášová, E.; Kovařík, A.; Kozubek, S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells 2018, 7, 11. https://doi.org/10.3390/cells7020011
Lukášová E, Kovařík A, Kozubek S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells. 2018; 7(2):11. https://doi.org/10.3390/cells7020011
Chicago/Turabian StyleLukášová, Emilie, Aleš Kovařík, and Stanislav Kozubek. 2018. "Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence" Cells 7, no. 2: 11. https://doi.org/10.3390/cells7020011
APA StyleLukášová, E., Kovařík, A., & Kozubek, S. (2018). Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells, 7(2), 11. https://doi.org/10.3390/cells7020011