MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages


Abstract
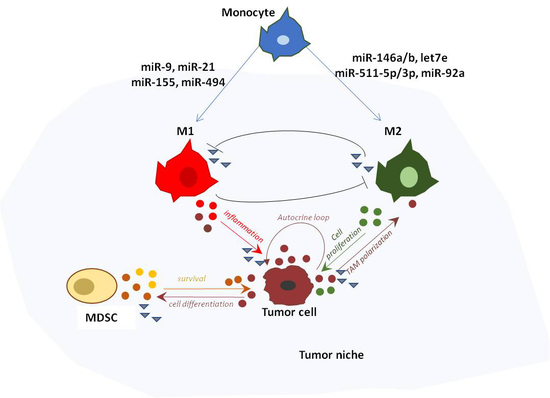
:
1. Introduction
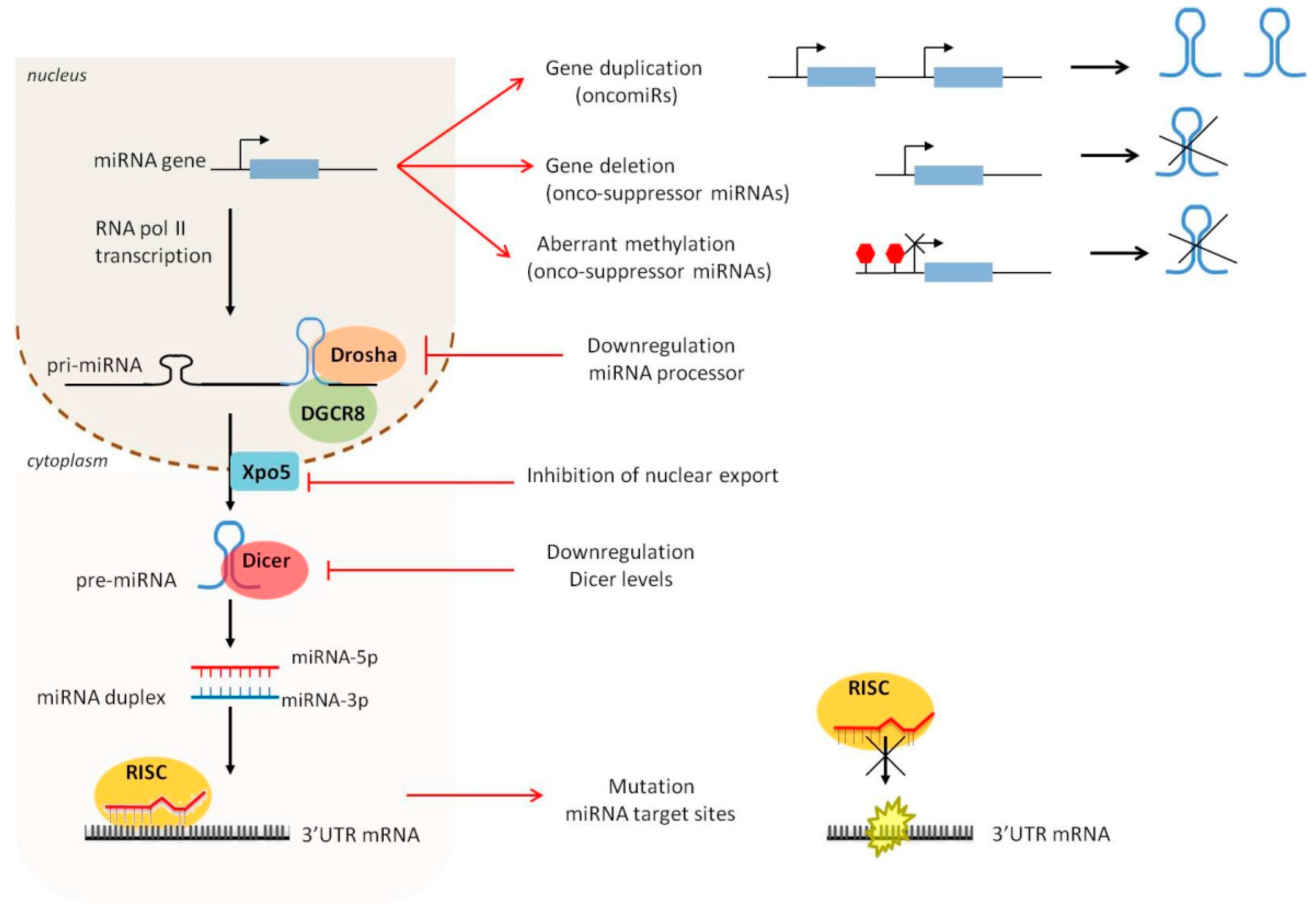
1.1. MiRNA Biogenesis and Maturation: A Brief Overview
1.2. Dysregulation of miRNA Expression and Biogenesis in Cancer
1.2.1. Amplification and Deletion of miRNA Genes
1.2.2. Epigenetic Alterations
1.2.3. Defects in the miRNA Biogenesis Pathway
1.2.4. Mutations of miRNA Target Genes
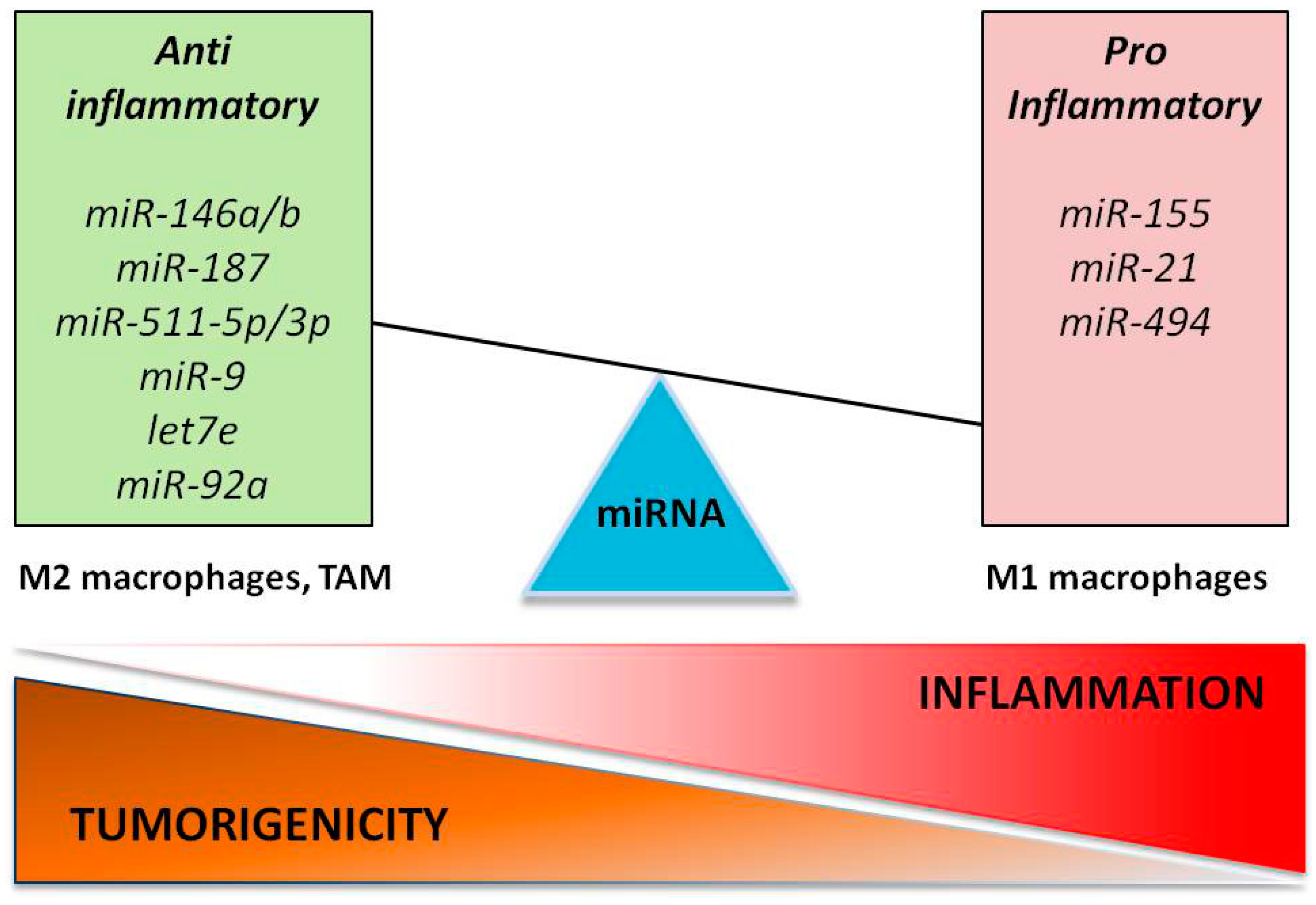
2. Role of miRNAs in Macrophage Polarization
3. The Fine Balance of the Innate Immune Tumor Response
4. miRNAs Involved in Tumor Immunosurveillance
5. TAM-Derived miRNAs in Cancer-Related Inflammation
6. Exosome-Derived miRNAs in Cancer Progression and Metastasis
7. Immunogenic Cell Death: A Promising Tool for Anti-Cancer Immunotherapy
miRNAs as Therapeutic Agents in Cancer Treatment
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bui, J.D.; Schreiber, R.D. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr. Opin. Immunol. 2007, 19, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Hagerling, C.; Casbon, A.J.; Werb, Z. Balancing the innate immune system in tumor development. Trends Cell Biol. 2015, 25, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Burnet, F.M. The concept of immunological surveillance. Prog. Exp. Tumor Res. 1970, 13, 1–27. [Google Scholar] [PubMed]
- Burnet, M. Cancer, a biological approach. I. The processes of control. Br. Med. J. 1957, 1, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M. Immunological Factors in the Process of Carcinogenesis. Br. Med. Bull. 1964, 20, 154–158. [Google Scholar] [CrossRef] [PubMed]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Street, S.E.; Cretney, E.; Smyth, M.J. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 2001, 97, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Trapani, J.A.; MacGregor, D.; Smyth, M.J. Suppression of lymphoma and epithelial malignancies effected by interferon gamma. J. Exp. Med. 2002, 196, 129–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinkai, Y.; Rathbun, G.; Lam, K.P.; Oltz, E.M.; Stewart, V.; Mendelsohn, M.; Charron, J.; Datta, M.; Young, F.; Stall, A.M.; et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Thia, K.Y.; Street, S.E.; MacGregor, D.; Godfrey, D.I.; Trapani, J.A. Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J. Exp. Med. 2000, 192, 755–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sanchez-Cespedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Caudy, A.A.; Hannon, G.J. Post-transcriptional gene silencing by double-stranded RNA. Nat. Rev. Genet. 2001, 2, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef]
- Kirigin, F.F.; Lindstedt, K.; Sellars, M.; Ciofani, M.; Low, S.L.; Jones, L.; Bell, F.; Pauli, F.; Bonneau, R.; Myers, R.M.; et al. Dynamic microRNA gene transcription and processing during T cell development. J. Immunol. 2012, 188, 3257–3267. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Kim, B.; Kim, V.N. Re-evaluation of the roles of DROSHA, Export in 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E1881–E1889. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Yan, K.P.; Amuthan, G.; Chendrimada, T.; Doratotaj, B.; Cooch, N.; Shiekhattar, R. The Microprocessor complex mediates the genesis of microRNAs. Nature 2004, 432, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Triboulet, R.; Chang, H.M.; Lapierre, R.J.; Gregory, R.I. Post-transcriptional control of DGCR8 expression by the Microprocessor. RNA 2009, 15, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, M.T.; Czaplinski, K.; Gorlich, D. Exportin 5 is a RanGTP-dependent dsRNA-binding protein that mediates nuclear export of pre-miRNAs. RNA 2004, 10, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Dahlberg, J.E. Substrate selectivity of exportin 5 and Dicer in the biogenesis of microRNAs. Cold Spring Harb. Symp. Quant. Biol. 2006, 71, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Grishok, A.; Pasquinelli, A.E.; Conte, D.; Li, N.; Parrish, S.; Ha, I.; Baillie, D.L.; Fire, A.; Ruvkun, G.; Mello, C.C. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell 2001, 106, 23–34. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef] [PubMed]
- De Moor, C.H.; Meijer, H.; Lissenden, S. Mechanisms of translational control by the 3’ UTR in development and differentiation. Semin. Cell Dev. Biol. 2005, 16, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Emoto, N.; Hamano, E.; Sunohara, M.; Kawakami, M.; Kage, H.; Kitano, K.; Nakajima, J.; Goto, A.; Fukayama, M.; et al. Genome structure-based screening identified epigenetically silenced microRNA associated with invasiveness in non-small-cell lung cancer. Int. J. Cancer 2012, 130, 2580–2590. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Guryev, V.; van de Belt, J.; Wienholds, E.; Plasterk, R.H.; Cuppen, E. Phylogenetic shadowing and computational identification of human microRNA genes. Cell 2005, 120, 21–24. [Google Scholar] [CrossRef] [PubMed]
- Boffelli, D.; McAuliffe, J.; Ovcharenko, D.; Lewis, K.D.; Ovcharenko, I.; Pachter, L.; Rubin, E.M. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 2003, 299, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivdasani, R.A. MicroRNAs: Regulators of gene expression and cell differentiation. Blood 2006, 108, 3646–3653. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Yamamoto, E.; Nojima, M.; Kai, M.; Yamano, H.O.; Yoshikawa, K.; Kimura, T.; Kudo, T.; Harada, E.; Sugai, T.; et al. Methylation-associated silencing of microRNA-34b/c in gastric cancer and its involvement in an epigenetic field defect. Carcinogenesis 2010, 31, 2066–2073. [Google Scholar] [CrossRef] [PubMed]
- Agirre, X.; Vilas-Zornoza, A.; Jimenez-Velasco, A.; Martin-Subero, J.I.; Cordeu, L.; Garate, L.; San Jose-Eneriz, E.; Abizanda, G.; Rodriguez-Otero, P.; Fortes, P.; et al. Epigenetic silencing of the tumor suppressor microRNA Hsa-miR-124a regulates CDK6 expression and confers a poor prognosis in acute lymphoblastic leukemia. Cancer Res. 2009, 69, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Volinia, S.; Calin, G.A.; Liu, C.G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, L.; Zhang, J.; Chen, H.; Fan, J.; Wang, K.; Luo, J.; Chen, Z.; Meng, Z.; Liu, L. Methylation-mediated silencing of the miR-124 genes facilitates pancreatic cancer progression and metastasis by targeting Rac1. Oncogene 2014, 33, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.Y.; So, C.C.; Loong, F.; Chung, L.P.; Lam, W.W.; Liang, R.; Li, G.K.; Jin, D.Y.; Chim, C.S. Epigenetic inactivation of the miR-124-1 in haematological malignancies. PLoS ONE 2011, 6, e19027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, M.S.; Lu, J.; Mercer, K.L.; Golub, T.R.; Jacks, T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat. Genet. 2007, 39, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Sevignani, C.; Calin, G.A.; Nnadi, S.C.; Shimizu, M.; Davuluri, R.V.; Hyslop, T.; Demant, P.; Croce, C.M.; Siracusa, L.D. MicroRNA genes are frequently located near mouse cancer susceptibility loci. Proc. Natl. Acad. Sci. USA 2007, 104, 8017–8022. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Hernandez-Herrera, A.; Jacobsen, A.; Levine, D.A.; Mankoo, P.; Schultz, N.; Du, Y.; Zhang, Y.; Larsson, E.; Sheridan, R.; et al. Integrated analyses of microRNAs demonstrate their widespread influence on gene expression in high-grade serous ovarian carcinoma. PLoS ONE 2012, 7, e34546. [Google Scholar] [CrossRef] [PubMed]
- Bayani, J.; Kuzmanov, U.; Saraon, P.; Fung, W.A.; Soosaipillai, A.; Squire, J.A.; Diamandis, E.P. Copy number and expression alterations of miRNAs in the ovarian cancer cell line OVCAR-3: Impact on kallikrein 6 protein expression. Clin. Chem. 2013, 59, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Imam, J.S.; Plyler, J.R.; Bansal, H.; Prajapati, S.; Bansal, S.; Rebeles, J.; Chen, H.I.; Chang, Y.F.; Panneerdoss, S.; Zoghi, B.; et al. Genomic loss of tumor suppressor miRNA-204 promotes cancer cell migration and invasion by activating AKT/mTOR/Rac1 signaling and actin reorganization. PLoS ONE 2012, 7, e52397. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.; Stresemann, C.; Brueckner, B.; Lyko, F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 2007, 6, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Muller, M.; Romermann, D.; Langer, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J. Pathol. 2008, 214, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Scott, G.K.; Mattie, M.D.; Berger, C.E.; Benz, S.C.; Benz, C.C. Rapid alteration of microRNA levels by histone deacetylase inhibition. Cancer Res. 2006, 66, 1277–1281. [Google Scholar] [CrossRef] [PubMed]
- Wee, E.J.; Peters, K.; Nair, S.S.; Hulf, T.; Stein, S.; Wagner, S.; Bailey, P.; Lee, S.Y.; Qu, W.J.; Brewster, B.; et al. Mapping the regulatory sequences controlling 93 breast cancer-associated miRNA genes leads to the identification of two functional promoters of the Hsa-mir-200b cluster, methylation of which is associated with metastasis or hormone receptor status in advanced breast cancer. Oncogene 2012, 31, 4182–4195. [Google Scholar] [CrossRef] [PubMed]
- Bandres, E.; Agirre, X.; Bitarte, N.; Ramirez, N.; Zarate, R.; Roman-Gomez, J.; Prosper, F.; Garcia-Foncillas, J. Epigenetic regulation of microRNA expression in colorectal cancer. Int. J. Cancer 2009, 125, 2737–2743. [Google Scholar] [CrossRef] [PubMed]
- Roman-Gomez, J.; Agirre, X.; Jimenez-Velasco, A.; Arqueros, V.; Vilas-Zornoza, A.; Rodriguez-Otero, P.; Martin-Subero, I.; Garate, L.; Cordeu, L.; San Jose-Eneriz, E.; et al. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, K.; Peters, I.; Dubrowinskaja, N.; Hennenlotter, J.; Abbas, M.; Scherer, R.; Tezval, H.; Merseburger, A.S.; Stenzl, A.; Kuczyk, M.A.; et al. Hsa-mir-124-3 CpG island methylation is associated with advanced tumours and disease recurrence of patients with clear cell renal cell carcinoma. Br. J. Cancer 2013, 108, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Furuta, M.; Kozaki, K.I.; Tanaka, S.; Arii, S.; Imoto, I.; Inazawa, J. miR-124 and miR-203 are epigenetically silenced tumor-suppressive microRNAs in hepatocellular carcinoma. Carcinogenesis 2010, 31, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010, 18, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Ropero, S.; Moutinho, C.; Aaltonen, L.A.; Yamamoto, H.; Calin, G.A.; Rossi, S.; Fernandez, A.F.; Carneiro, F.; Oliveira, C.; et al. A TARBP2 mutation in human cancer impairs microRNA processing and DICER1 function. Nat. Genet. 2009, 41, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.S.; Pester, R.E.; Chen, C.Y.; Lane, K.; Chin, C.; Lu, J.; Kirsch, D.G.; Golub, T.R.; Jacks, T. Dicer1 functions as a haploinsufficient tumor suppressor. Genes Dev. 2009, 23, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Garibaldi, F.; Falcone, E.; Trisciuoglio, D.; Colombo, T.; Lisek, K.; Walerych, D.; Del Sal, G.; Paci, P.; Bossi, G.; Piaggio, G.; et al. Mutant p53 inhibits miRNA biogenesis by interfering with the microprocessor complex. Oncogene 2016, 35, 3760–3770. [Google Scholar] [CrossRef] [PubMed]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, C.; Hemann, M.T.; Bartel, D.P. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007, 315, 1576–1579. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Dutta, A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007, 21, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.J.; Ratner, E.; Leng, S.; Zhai, R.; Nallur, S.; Babar, I.; Muller, R.U.; Straka, E.; Su, L.; Burki, E.A.; et al. A SNP in a let-7 microRNA complementary site in the KRAS 3′ untranslated region increases non-small cell lung cancer risk. Cancer Res. 2008, 68, 8535–8540. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, M.; Zhong, M.; Suo, Q.; Lv, K. Expression profiles of miRNAs in polarized macrophages. Int. J. Mol. Med. 2013, 31, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Abraham, E. MicroRNAs in immune response and macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Cobos Jimenez, V.; Bradley, E.J.; Willemsen, A.M.; van Kampen, A.H.; Baas, F.; Kootstra, N.A. Next-generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol. Genom. 2014, 46, 91–103. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Chaudhuri, A.A.; Rao, D.S.; Baltimore, D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc. Natl. Acad. Sci. USA 2009, 106, 7113–7118. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014, 124, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Singh, U.P.; Murphy, A.E.; Enos, R.T.; Shamran, H.A.; Singh, N.P.; Guan, H.; Hegde, V.L.; Fan, D.; Price, R.L.; Taub, D.D.; et al. miR-155 deficiency protects mice from experimental colitis by reducing T helper type 1/type 17 responses. Immunology 2014, 143, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Nunez, R.T.; Louafi, F.; Sanchez-Elsner, T. The interleukin 13 (IL-13) pathway in human macrophages is modulated by microRNA-155 via direct targeting of interleukin 13 receptor alpha1 (IL13Ralpha1). J. Biol. Chem. 2011, 286, 1786–1794. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.A.; So, A.Y.; Sinha, N.; Gibson, W.S.; Taganov, K.D.; O’Connell, R.M.; Baltimore, D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011, 187, 5062–5068. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Duan, M.; Feng, Y.; Geng, L.; Li, X.; Zhang, W. MiR-146a modulates macrophage polarization in systemic juvenile idiopathic arthritis by targeting INHBA. Mol. Immunol. 2016, 77, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, X.J.; Xie, J.; Ma, T.T.; Meng, X.M.; Li, J. MiR-146a modulates macrophage polarization by inhibiting Notch1 pathway in RAW264.7 macrophages. Int. Immunopharmacol. 2016, 32, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; O’Neill, L.A.; Masters, S.L. miR-223: Infection, inflammation and cancer. J. Int. Med. 2013, 274, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA let-7c regulates macrophage polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.; Liu, W.; Liu, Y.; Xu, J. Polycomb-mediated loss of microRNA let-7c determines inflammatory macrophage polarization via PAK1-dependent NF-kappaB pathway. Cell Death Differ. 2015, 22, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Holdbrooks, A.T.; Liu, Y.; Reynolds, S.L.; Yanagisawa, L.L.; Benveniste, E.N. Correction: SOCS3 Deficiency Promotes M1 Macrophage Polarization and Inflammation. J. Immunol. 2016, 197, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Zhang, P.; Liang, T.; Deng, S.; Chen, X.; Zhu, L. Ovarian cancer stem cells induce the M2 polarization of macrophages through the PPARgamma and NF-kappaB pathways. Int. J. Mol. Med. 2015, 36, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.G.; Freemont, P.S.; Foulkes, W.; Trowsdale, J. An ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains. Cancer Res. 1992, 52, 5416–5420. [Google Scholar] [PubMed]
- Tsai, R.K.; Discher, D.E. Inhibition of “self“ engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D., Jr.; van Rooijen, N.; Weissman, I.L. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Topfer, K.; Kempe, S.; Muller, N.; Schmitz, M.; Bachmann, M.; Cartellieri, M.; Schackert, G.; Temme, A. Tumor evasion from T cell surveillance. J. Biomed. Biotechnol. 2011, 2011, 918471. [Google Scholar] [CrossRef] [PubMed]
- Waldhauer, I.; Steinle, A. NK cells and cancer immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef] [PubMed]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Squadrito, M.L.; Pucci, F.; Magri, L.; Moi, D.; Gilfillan, G.D.; Ranghetti, A.; Casazza, A.; Mazzone, M.; Lyle, R.; Naldini, L.; et al. miR-511-3p modulates genetic programs of tumor-associated macrophages. Cell Rep. 2012, 1, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Stockmann, C.; Rubinstein, M.P.; Liao, D.; Zhang, N.; DeNardo, D.G.; Coussens, L.M.; Karin, M.; Goldrath, A.W.; Johnson, R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010, 70, 7465–7475. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Gabrilovich, D.I. Molecular pathways: Tumor-infiltrating myeloid cells and reactive oxygen species in regulation of tumor microenvironment. Clin. Cancer Res. 2012, 18, 4877–4882. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.I.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.B.; Gabrilovich, D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Investig. 2011, 121, 4015–4029. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.K.; Kim, J.S.; Lee, C.G.; Park, Y.S.; Kim, S.D.; Yoon, S.O.; Han, D.H.; Lee, K.Y.; Jeong, M.H.; Jo, W.S. Tumor associated macrophages provide the survival resistance of tumor cells to hypoxic microenvironmental condition through IL-6 receptor-mediated signals. Immunobiology 2017, 222, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, J.; Chen, S.; Lu, M.; Luo, X.; Yao, S.; Liu, S.; Qin, Y.; Chen, H. Tumor-associated macrophages provide a suitable microenvironment for non-small lung cancer invasion and progression. Lung Cancer 2011, 74, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Mastio, J.; Gabrilovich, D.I. Transcriptional regulation of myeloid-derived suppressor cells. J. Leukoc. Biol. 2015, 98, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Filipazzi, P.; Huber, V.; Rivoltini, L. Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. CII 2012, 61, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gabrilovich, D.I. Tumor escape mechanism governed by myeloid-derived suppressor cells. Cancer Res. 2008, 68, 2561–2563. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Reciprocal relationship between myeloid-derived suppressor cells and T cells. J. Immunol. 2013, 191, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.M.; Clements, V.K.; Sinha, P.; Ilkovitch, D.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J. Immunol. 2009, 183, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, J.; Ren, J.P.; Wu, X.Y.; Morrison, Z.D.; Elgazzar, M.A.; Ning, S.B.; Moorman, J.P.; Yao, Z.Q. Expansion of myeloid-derived suppressor cells promotes differentiation of regulatory T cells in HIV-1+ individuals. Aids 2016, 30, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. Myeloid-derived suppressor cells regulate natural killer cell response to adenovirus-mediated gene transfer. J. Virol. 2012, 86, 13689–13696. [Google Scholar] [CrossRef] [PubMed]
- Hoechst, B.; Voigtlaender, T.; Ormandy, L.; Gamrekelashvili, J.; Zhao, F.; Wedemeyer, H.; Lehner, F.; Manns, M.P.; Greten, T.F.; Korangy, F. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 2009, 50, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Du, W.; Yan, F.; Wang, Y.; Li, H.; Cao, S.; Yu, W.; Shen, C.; Liu, J.; Ren, X. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 2013, 190, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, M.L.; Lu, L.; Ramachandran, I.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the development of lung cancer. Cancer Immunol. Res. 2014, 2, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Goldszmid, R.S.; Muranski, P.; Chinnasamy, D.; Yu, Z.; Reger, R.N.; Leonardi, A.J.; Morgan, R.A.; Wang, E.; Marincola, F.M.; et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J. Clin. Investig. 2011, 121, 4746–4757. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Hua, L.M.; Guo, M.; Luo, J.M. SHIP1 is targeted by miR-155 in acute myeloid leukemia. Oncol. Rep. 2014, 32, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Fan, J.; Ye, C.; Dominguez, D.; Zhang, Y.; Curiel, T.J.; Fang, D.; Kuzel, T.M.; Zhang, B. Host miR155 promotes tumor growth through a myeloid-derived suppressor cell-dependent mechanism. Cancer Res. 2015, 75, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Cantoni, C.; Cignarella, F.; Ghezzi, L.; Mikesell, B.; Bollman, B.; Berrien-Elliott, M.M.; Ireland, A.R.; Fehniger, T.A.; Wu, G.F.; Piccio, L. Mir-223 regulates the number and function of myeloid-derived suppressor cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2017, 133, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhang, M.; Jiang, X.; Zhang, Z.; Dai, L.; Min, S.; Wu, X.; He, Q.; Liu, J.; Zhang, Y.; et al. miR-223 suppresses differentiation of tumor-induced CD11b(+) Gr1(+) myeloid-derived suppressor cells from bone marrow cells. Int. J. Cancer 2011, 129, 2662–2673. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.M.; Ramos, K.S.; Colburn, N.H.; Li, Y. MicroRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef] [PubMed]
- Hegde, V.L.; Tomar, S.; Jackson, A.; Rao, R.; Yang, X.; Singh, U.P.; Singh, N.P.; Nagarkatti, P.S.; Nagarkatti, M. Distinct microRNA expression profile and targeted biological pathways in functional myeloid-derived suppressor cells induced by Delta9-tetrahydrocannabinol in vivo: Regulation of CCAAT/enhancer-binding protein alpha by microRNA-690. J. Biol. Chem. 2013, 288, 36810–36826. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol. 2009, 86, 1065–1073. [Google Scholar] [CrossRef] [PubMed]
- Igaz, I.; Igaz, P. Are Circulating microRNAs Involved in Tumor Surveillance? In Circulating microRNAs in Disease Diagnostics and their Potential Biological Relevance; Springer: Basel, Switzerland, 2015; Volume 106, pp. 269–280. [Google Scholar] [CrossRef] [PubMed]
- Nandi, B.; Shapiro, M.; Samur, M.K.; Pai, C.; Frank, N.Y.; Yoon, C.; Prabhala, R.H.; Munshi, N.C.; Gold, J.S. Stromal CCR6 drives tumor growth in a murine transplantable colon cancer through recruitment of tumor-promoting macrophages. Oncoimmunology 2016, 5, e1189052. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Wu, C.C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, Y.; Wang, N.; Zhang, X.; Tan, B.; Zhang, G.; Cheng, Y. The clinical significance of tumor-infiltrating neutrophils and neutrophil-to-CD8+ lymphocyte ratio in patients with resectable esophageal squamous cell carcinoma. J. Transl. Med. 2014, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Zou, Y.; He, X.; Yuan, R.; Chen, Y.; Lan, N.; Lian, L.; Wang, F.; Fan, X.; Zeng, Y.; et al. Tumor-infiltrating mast cells in colorectal cancer as a poor prognostic factor. Int. J. Surg. Pathol. 2013, 21, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Zhu, Y.; Wang, Y.; Liu, Z.; Zhang, J.; Wang, Z.; Xie, H.; Dai, B.; Xu, J.; Ye, D. Tumor Infiltrating Mast Cells (TIMs) Confers a Marked Survival Advantage in Nonmetastatic Clear-Cell Renal Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Renzi, T.A.; Rubino, M.; Gornati, L.; Garlanda, C.; Locati, M.; Curtale, G. MiR-146b Mediates Endotoxin Tolerance in Human Phagocytes. Mediat. Inflamm. 2015, 2015, 145305. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Wen, X.Y.; Yang, H.K.; Kim, W.H.; Kang, G.H. Prognostic Implication of M2 Macrophages Are Determined by the Proportional Balance of Tumor Associated Macrophages and Tumor Infiltrating Lymphocytes in Microsatellite-Unstable Gastric Carcinoma. PLoS ONE 2015, 10, e0144192. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; DeGuzman, A.; Bucana, C.D.; Fidler, I.J. Nuclear factor-kappaB activity correlates with growth, angiogenesis, and metastasis of human melanoma cells in nude mice. Clin. Cancer Res. 2000, 6, 2573–2581. [Google Scholar] [PubMed]
- Cekaite, L.; Rantala, J.K.; Bruun, J.; Guriby, M.; Agesen, T.H.; Danielsen, S.A.; Lind, G.E.; Nesbakken, A.; Kallioniemi, O.; Lothe, R.A.; et al. MiR-9, -31, and -182 deregulation promote proliferation and tumor cell survival in colon cancer. Neoplasia 2012, 14, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasier, A.R. The nuclear factor-kappaB-interleukin-6 signalling pathway mediating vascular inflammation. Cardiovasc. Res. 2010, 86, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Masuda, T.; Iinuma, H.; Yamaguchi, R.; Sato, K.; Tobo, T.; Hirata, H.; Kuroda, Y.; Nambara, S.; Hayashi, N.; et al. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget 2017, 8, 78598–78613. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. CR 2017, 36, 53. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.A.; Gooderham, N.J. IL6 Mediates Immune and Colorectal Cancer Cell Cross-talk via miR-21 and miR-29b. Mol. Cancer Res. MCR 2015, 13, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Curtale, G.; Renzi, T.A.; Drufuca, L.; Rubino, M.; Locati, M. Glucocorticoids downregulate TLR4 signaling activity via its direct targeting by miR-511-5p. Eur. J. Immunol. 2017, 47, 2080–2089. [Google Scholar] [CrossRef] [PubMed]
- Rossato, M.; Curtale, G.; Tamassia, N.; Castellucci, M.; Mori, L.; Gasperini, S.; Mariotti, B.; De Luca, M.; Mirolo, M.; Cassatella, M.A.; et al. IL-10-induced microRNA-187 negatively regulates TNF-alpha, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, E3101–E3110. [Google Scholar] [CrossRef] [PubMed]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Song, Y.; Liu, Y.; Chen, Q.; Han, Q.; Chen, W.; Pan, T.; Zhang, Y.; Cao, X.; Wang, Q. MicroRNA-92a negatively regulates Toll-like receptor (TLR)-triggered inflammatory response in macrophages by targeting MKK4 kinase. J. Biol. Chem. 2013, 288, 7956–7967. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Liu, Y.; McFarland, B.C.; Deshane, J.S.; Hurst, D.R.; Ponnazhagan, S.; Benveniste, E.N.; Qin, H. SOCS3 Deficiency in Myeloid Cells Promotes Tumor Development: Involvement of STAT3 Activation and Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2015, 3, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Liu, Q.; Mi, S.; Liang, X.; Zhang, Z.; Su, X.; Liu, J.; Chen, Y.; Wang, M.; Zhang, Y.; et al. Both miR-17-5p and miR-20a alleviate suppressive potential of myeloid-derived suppressor cells by modulating STAT3 expression. J. Immunol. 2011, 186, 4716–4724. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Gomis, R.R. TGF-beta Family Signaling in Tumor Suppression and Cancer Progression. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Soon, P.; Kiaris, H. MicroRNAs in the tumour microenvironment: Big role for small players. Endocr.-Relat. Cancer 2013, 20, R257–R267. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ying, X.; Wang, X.; Wu, X.; Zhu, Q.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol. Rep. 2017, 38, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Imaoka, H.; Toiyama, Y.; Okigami, M.; Yasuda, H.; Saigusa, S.; Ohi, M.; Tanaka, K.; Inoue, Y.; Mohri, Y.; Kusunoki, M. Circulating microRNA-203 predicts metastases, early recurrence, and poor prognosis in human gastric cancer. Gastric Cancer Off. J. Int. Gastric Cancer Assoc. Jpn. Gastric Cancer Assoc. 2016, 19, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Toiyama, Y.; Okugawa, Y.; Ide, S.; Imaoka, H.; Boland, C.R.; Goel, A. Circulating microRNA-203 predicts prognosis and metastasis in human colorectal cancer. Gut 2017, 66, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Rask, L.; Balslev, E.; Jorgensen, S.; Eriksen, J.; Flyger, H.; Moller, S.; Hogdall, E.; Litman, T.; Nielsen, B.S. High expression of miR-21 in tumor stroma correlates with increased cancer cell proliferation in human breast cancer. APMIS: Acta Pathol. Microbiol. Immunol. Scand. 2011, 119, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.X.; Wu, Q.N.; Zhang, Y.; Li, Y.Y.; Liao, D.Z.; Hou, J.H.; Fu, J.; Zeng, M.S.; Yun, J.P.; Wu, Q.L.; et al. Knockdown of miR-21 in human breast cancer cell lines inhibits proliferation, in vitro migration and in vivo tumor growth. Breast Cancer Res. BCR 2011, 13, R2. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.J.; Foersch, S.; Neurath, M.F. Interleukin-6--a key regulator of colorectal cancer development. Int. J. Biol. Sci. 2012, 8, 1248–1253. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, G.; Wu, X.; Jiang, Z.; Kasman, I.; Yao, J.; Guan, Y.; Oeh, J.; Modrusan, Z.; Bais, C.; Sampath, D.; et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J. 2012, 31, 3513–3523. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, C.; Yang, D.; Liao, Q.; Luo, H.; Wang, X.; Zhou, F.; Yang, X.; Yang, J.; Zeng, C.; et al. Mesenchymal stem cells-derived extracellular vesicles, via miR-210, improve infarcted cardiac function by promotion of angiogenesis. Biochim. Biophys. Acta 2017, 1863, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Le, M.T.; Hamar, P.; Guo, C.; Basar, E.; Perdigao-Henriques, R.; Balaj, L.; Lieberman, J. miR-200-containing extracellular vesicles promote breast cancer cell metastasis. J. Clin. Investig. 2014, 124, 5109–5128. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Salomon, C.; Tapia, J.; Illanes, S.E.; Mitchell, M.D.; Rice, G.E. Ovarian cancer cell invasiveness is associated with discordant exosomal sequestration of Let-7 miRNA and miR-200. J. Transl. Med. 2014, 12, 4. [Google Scholar] [CrossRef] [PubMed]
- Epstein, D.M. Special delivery: MicroRNA-200-containing extracellular vesicles provide metastatic message to distal tumor cells. J. Clin. Investig. 2014, 124, 5107–5108. [Google Scholar] [CrossRef] [PubMed]
- Kanlikilicer, P.; Rashed, M.H.; Bayraktar, R.; Mitra, R.; Ivan, C.; Aslan, B.; Zhang, X.; Filant, J.; Silva, A.M.; Rodriguez-Aguayo, C.; et al. Ubiquitous Release of Exosomal Tumor Suppressor miR-6126 from Ovarian Cancer Cells. Cancer Res. 2016, 76, 7194–7207. [Google Scholar] [CrossRef] [PubMed]
- Qattan, A.; Intabli, H.; Alkhayal, W.; Eltabache, C.; Tweigieri, T.; Amer, S.B. Robust expression of tumor suppressor miRNA’s let-7 and miR-195 detected in plasma of Saudi female breast cancer patients. BMC Cancer 2017, 17, 799. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Kang, Y.; He, Y.; Liu, J.; Liang, B.; Yang, P.; Yu, Z. microRNA-99a acts as a tumor suppressor and is down-regulated in bladder cancer. BMC Urol. 2014, 14, 50. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.; Yamaguchi, K.; et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 2010, 5, e13247. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Nishikawa, R.; Kojima, S.; Chiyomaru, T.; Enokida, H.; Inoguchi, S.; Kinoshita, T.; Fuse, M.; Sakamoto, S.; Nakagawa, M.; et al. Tumour-suppressive microRNA-224 inhibits cancer cell migration and invasion via targeting oncogenic TPD52 in prostate cancer. FEBS Lett. 2014, 588, 1973–1982. [Google Scholar] [CrossRef] [PubMed]
- Ostenfeld, M.S.; Jeppesen, D.K.; Laurberg, J.R.; Boysen, A.T.; Bramsen, J.B.; Primdal-Bengtson, B.; Hendrix, A.; Lamy, P.; Dagnaes-Hansen, F.; Rasmussen, M.H.; et al. Cellular disposal of miR23b by RAB27-dependent exosome release is linked to acquisition of metastatic properties. Cancer Res. 2014, 74, 5758–5771. [Google Scholar] [CrossRef] [PubMed]
- Donadelli, M.; Dando, I.; Fiorini, C.; Palmieri, M. Regulation of miR-23b expression and its dual role on ROS production and tumour development. Cancer Lett. 2014, 349, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Igaz, I.; Igaz, P. Tumor surveillance by circulating microRNAs: A hypothesis. Cell. Mol. Life Sci. CMLS 2014, 71, 4081–4087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, A.M.; Garg, A.D.; Krysko, D.V.; De Ruysscher, D.; Agostinis, P. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013, 24, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Vandenabeele, P.; Vandecasteele, K.; Bachert, C.; Krysko, O.; Krysko, D.V. Immunogenic Apoptotic Cell Death and Anticancer Immunity. Adv. Exp. Med. Biol. 2016, 930, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Colangelo, T.; Polcaro, G.; Ziccardi, P.; Muccillo, L.; Galgani, M.; Pucci, B.; Milone, M.R.; Budillon, A.; Santopaolo, M.; Mazzoccoli, G.; et al. The miR-27a-calreticulin axis affects drug-induced immunogenic cell death in human colorectal cancer cells. Cell Death Dis. 2016, 7, e2108. [Google Scholar] [CrossRef] [PubMed]
- Alderton, G.K.; Bordon, Y. Tumour immunotherapy—Leukocytes take up the fight. Nat. Rev. Immunol. 2012, 12, 237. [Google Scholar] [CrossRef] [PubMed]
- Bird, L. Immunotherapy: Remote control CARs. Nat. Rev. Drug Discov. 2015, 14, 819. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Tseng, D.; Volkmer, J.P.; Willingham, S.B.; Contreras-Trujillo, H.; Fathman, J.W.; Fernhoff, N.B.; Seita, J.; Inlay, M.A.; Weiskopf, K.; Miyanishi, M.; et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. USA 2013, 110, 11103–11108. [Google Scholar] [CrossRef] [PubMed]
- Iclozan, C.; Antonia, S.; Chiappori, A.; Chen, D.T.; Gabrilovich, D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol. Immunother. CII 2013, 62, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Gul, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bogels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Dalmay, T.; Edwards, D.R. MicroRNAs and the hallmarks of cancer. Oncogene 2006, 25, 6170–6175. [Google Scholar] [CrossRef] [PubMed]
- Ruan, K.; Fang, X.; Ouyang, G. MicroRNAs: Novel regulators in the hallmarks of human cancer. Cancer Lett. 2009, 285, 116–126. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
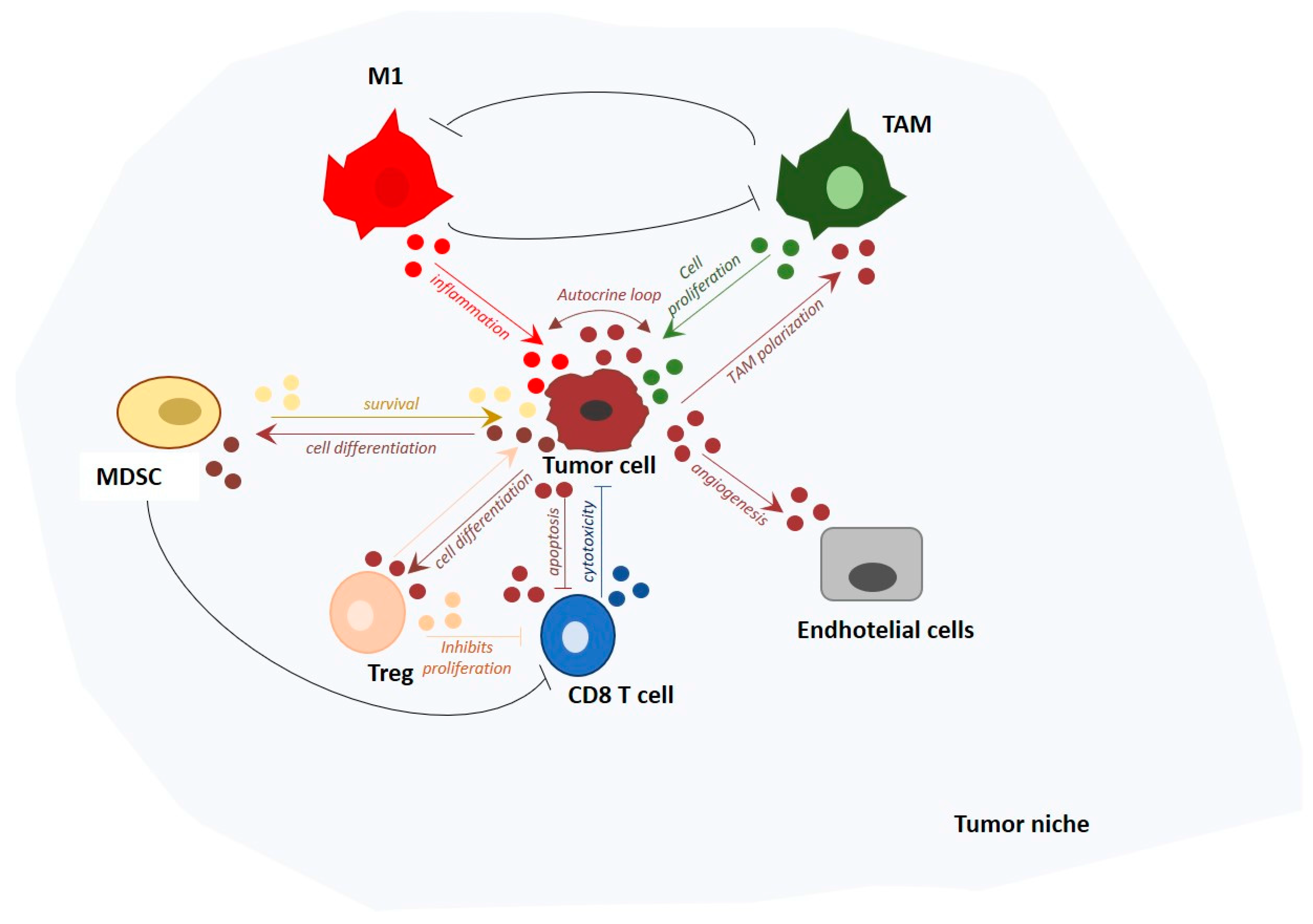{kind=link}
| miRNA | Expression | Targets | Phenotype | |
|---|---|---|---|---|
| miR-155 | ↑ M1 macrophages | C/EBPβ, SHIP1, IL13Rα1, SMAD2/3 | Reprograms pro-tumoral M2/TAM macrophages to M1 pro-inflammatory macrophages | Macrophage Polarization |
| miR-125b | ↑ M1 macrophages | IRF4 | ↑ responsiveness to IFNγ ↑ tumor killing | |
| miR-127 | ↑ in M2 macrophages ↓ by inflammation | DUSP1 | ↑ M1- and ↓ M2-related genes | |
| miR-146a | ↑ M2 macrophages | NOTCH1, INHBA, PPARγ, | ↑ M2 polarization and inflammation ↓ M1 polarization | |
| miR-223 | ↓ TAM | IL1β, IL-6 | ↑ M2 polarization | |
| let-7c | ↑ in M2 macrophages ↓ by inflammation | C/EBPδ, PAK1 | ↑ M2- and ↓ M1-related genes | |
| miR-511-3p | ↑ TAM | ROCK2 | ↓ pro-tumoral gene signature of TAMS and ↓ tumor growth | Tumor invasion |
| miR-155 | ↑ M1 macrophages | SHIP1 | ↑ anti-tumor immunity. MiR-155 KO myeloid cells induce faster tumor growth | |
| miR-155 | ↑ MDSC | SOCS1 | Required for tumor growth and the generation of CD4+ Treg cells. MiR-155 KO mice are resistant to carcinogenesis | Immune suppression (MDSC) |
| miR-494 | ↑ MDSC | PTEN | Regulates cell cycle progression; it induces arrest in G2/M and increased inflammation | |
| miR-20a | ↑ MDSC | STAT3 | ↓ MDSC-dependent suppression of CD4+ and CD8+ T cell response | |
| miR-223 | ↓ MDSC | MEF2C | Suppresses differentiation of tumor induced- CD11bGr1+MDSC | |
| miR-21 | ↑ MDSC | SHIP1 | ↑ proliferation and survival | |
| miR-690 | ↑ MDSC | C/EBPα | ↑ MDSC expansion and proliferation ↓ terminal differentiation | |
| miR-17-5p | ↑ MDSC | STAT3 | ↓ MDSC ability to suppress Ag-specific CD4+ and CD8+ T cell response |
| miRNA | Donor Cells | Recipient Cells | Phenotype | |
|---|---|---|---|---|
| miR-21 | TAM | gastric cancer cells | ↑ cell proliferation ↓ chemosensitivity and apoptosis | TAM-derived exosomal miRNAs |
| miR-223 | TAM | breast cancer cells | ↑ cell migration capacity and invasiveness | |
| miR-9 | TAM | endothelial cells | ↑ cell migration and angiogenesis | |
| miR-940 | epithelial ovarian cancer | TAM | ↑ M2 polarization | Tumor-secreted exosomal miRNAs |
| miR-203 | colorectal carcinoma | monocytes | ↑ M2 polarization | |
| miR-21 | solid tumor cells | macrophages | ↑ proliferation, survival, invasion and migration | |
| miR-29b | solid tumor cells | macrophages | OncomiR: ↑ secretion of pro-metastatic and pro-inflammatory cytokines | |
| miR-9 | tumor cell lines | cancer cells | OncomiR: ↑ endothelial cell migration ↑ tumor angiogenesis | |
| miR-210 | breast cancer cells | adjacent cancer cells | OncomiR: ↑ angiogenesis | |
| miR-105 | breast cancer cells | endothelial cells of distant organs | OncomiR: ↑ metastasis and vascular permeability by targeting the tight junction protein ZO-1 | |
| miR-200 | metastatic cancer cells | metastatic cancer cells | Regulates mesenchimal to epithelial transition | |
| let-7e | gastric cancer cells | cancer cells | Tumor suppressor function (i.e., inhibits metastasis) | |
| miR-23b | bladder carcinoma cells | cancer cells | Tumor suppressor function (e.g., inhibition angiogenesis, invasion and metastasis) | |
| miR-224 | bladder carcinoma cells | cancer cells | Tumor suppressor functions (e.g., inhibition angiogenesis, invasion and metastasis) | |
| miR-921 | bladder carcinoma cells | cancer cells | Tumor suppressor functions (e.g., inhibition angiogenesis, invasion and metastasis) | |
| miR-15a | mesenchymal stromal cells | myeloma cells | Tumor suppressor function |
| Immunological Target | Drug Name | Characteristics | Clinical Phase Testing | |
|---|---|---|---|---|
| Drugs blocking Immune Checkpoints | anti-CTLA4 | IPILIMUMAB | fully human IgG1 | approved, advanced melanoma |
| TREMELIMUMAB | fully human IgG2 | failed phase III trial melanoma | ||
| anti-PD1 | NIVOLUMAB | fully human IgG4 | approved, melanoma, squamous NSCLC | |
| PEMBROLIZUMAB | fully human IgG4 | approved, melanoma | ||
| PIDILIZUMAB | humanized IgG1 | phase I-II trial | ||
| anti-PD-L1 | BMS-936559 | fully human IgG4 | phase I trial | |
| MSB0010718C | fully human IgG1 | phase I-II trial | ||
| MEDI4736 | Fc-modified human IgG1 | phase I-III trial | ||
| MPDL3280A | Fc-modified human IgG1 | phase I-III trial | ||
| CAR T cells therapy | anti-CD19 | TISAGENLECLEUCEL-T (KimryahTM) | chimeric antigen receptor T cells | approved, Acute lymphatic leukemia |
| AXICABTAGENE CILOLEUCEL (YescartaTM) | chimeric antigen receptor T cells | approved, B cell lymphoma | ||
| anti-CD20 | NCT01735604 | chimeric antigen receptor T cells | phase I trial, progressive malignant lymphoma | |
| anti-CD30 | NCT02259556 | chimeric antigen receptor T cells | phase II-III trial, Hodgkin lymphoma | |
| anti-EGFR | NCT01869166 | chimeric antigen receptor T cells | phase II-III trial, Advanced lung cancer | |
| miRNA-based drugs | miR-34 | MRX34 | miRNA mimic | phase I trial, advanced hepatocarcinoma |
| miR-122 | MIRAVIRSEN | LNA-based antimiRNA | phase II trial, HCV | |
| miR-29b | MRG-201 | miRNA mimic | phase I trial, fibrotic diseases | |
| miR-155 | MRG-106 | AntimiRNA | phase I trial, hematological malignances | |
| miR-10b | - | AntimiRNA | preclinical phase, glioblastoma | |
| miR-221 | - | AntimiRNA | preclinical phase, hepatocarcinoma |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curtale, G. MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages. Cells 2018, 7, 12. https://doi.org/10.3390/cells7020012
Curtale G. MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages. Cells. 2018; 7(2):12. https://doi.org/10.3390/cells7020012
Chicago/Turabian StyleCurtale, Graziella. 2018. "MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages" Cells 7, no. 2: 12. https://doi.org/10.3390/cells7020012
APA StyleCurtale, G. (2018). MiRNAs at the Crossroads between Innate Immunity and Cancer: Focus on Macrophages. Cells, 7(2), 12. https://doi.org/10.3390/cells7020012