Linear Regression QSAR Models for Polo-Like Kinase-1 Inhibitors

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Experimental Dataset
2.2. Structural Representation and Molecular Descriptors Calculation
2.3. Model Development
2.3.1. Molecular Descriptors Selection
2.3.2. Model Validation
2.3.3. Applicability Domain
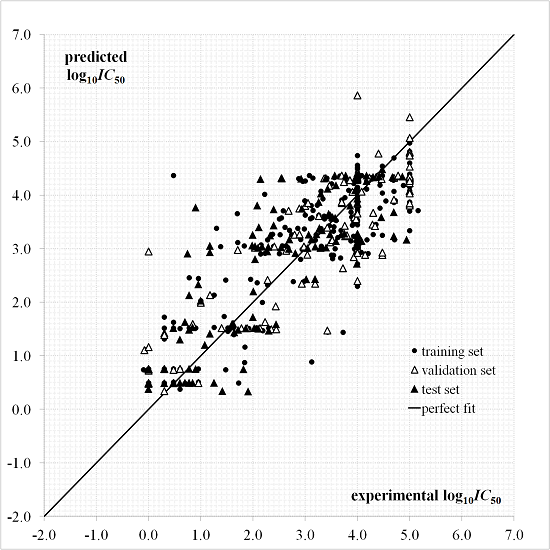
3. Results and Discussion
- Two electrotopological state atom-type descriptors: mindssC, the minimum atom-type E-state: =C<; and maxHCsats, the maximum atom-type H E-state: H bonded to B, Si, P, Ge, As, Se, Sn, or Pb.
- A MACCS fingerprint descriptor: M66, the number of CC(C)(C)A fragments, where A is any valid periodic table element symbol.
- Three PubChem fingerprint descriptors: PC494, the presence of O=C-C:N fragment, where ‘:’ denotes bond aromaticity; PC534, the presence of S-C:C-O fragment; and PC686, the presence of O=C-C-C-C-O fragment.
- Two Klekota–Roth fingerprint descriptors: KR3577, the presence of SMARTS substructure Cc1cccc(C)c1NC=O; and KR4268, the presence of SMARTS substructure Nc1ccccc1O.
- i.
- Our proposed model performs both regression and classification.
- ii.
- Dataset partitioning: three subsets are considered, such as train, val, and test instead of only two (train and test) in [20]. In this way, it is more convenient for analyzing the predictive performance of the model.
- iii.
- Model’s size: a fewer number of molecular descriptors are involved in the final selected model—i.e., 8 instead of 10–15. Therefore, the parsimony´s principle is accomplished (Ockham’s razor) [49] by following the common practice of keeping the model’s dimension as small as possible.
- iv.
- No energy or geometry optimization is performed on the inhibitor chemical structures. The conformation-independent QSAR approach considers only constitutional and topological representations for deriving the molecular descriptors.
- v.
- A simpler modeling methodology based on MLR analysis is applied in the present study.
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Lowery, D.M.; Lim, D.; Yaffe, M.B. Structure and function of Polo-like kinases. Oncogene 2005, 24, 248–259. [Google Scholar] [CrossRef] [PubMed]
- McInnes, C.; Wyatt, M.D. PLK1 as an oncology target: Current status and future potential. Drug Discov. Today 2011, 16, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Shakil, S.; Baig, M.H.; Tabrez, S.; Danish Rizvi, S.M.; Zaidi, S.K.; Ashraf, G.M.; Ansari, S.A.; Parwaz Khan, A.A.; Al-Qahtani, M.H.; Abuzenadah, A.M.; et al. Molecular and enzoinformatics perspectives of targeting Polo-like kinase 1 in cancer therapy. Semin. Cancer Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Burke, T.R., Jr.; Park, J.E.; Bang, J.K.; Lee, E. Recent Advances and New Strategies in Targeting PLK1 for Anticancer Therapy. Trends Pharmacol. Sci. 2015, 36, 858–877. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Becker, S.; Matthess, Y. Thoughts on the current assessment of Polo-like kinase inhibitor drug discovery. Expert Opin. Drug Discov. 2015, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, P.Y.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Chopra, P.; Sethi, G.; Dastidar, S.G.; Ray, A. Polo-like kinase inhibitors: An emerging opportunity for cancer therapeutics. Expert Opin. Investig. Drugs 2010, 19, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Leo, A. Exploring QSAR. Fundamentals and Applications in Chemistry and Biology; American Chemical Society: Washington, DC, USA, 1995. [Google Scholar]
- Esposito, E.; Hopfinger, A.J. Multi-Dimensional QSAR: Methods and Applications for Drug Discovery and Polymer Science; CRC Press: New York, NY, USA, 2012. [Google Scholar]
- Putz, M.V. QSAR and SPECTRAL-SAR in Computational Ecotoxicology; CRC Press: New York, NY, USA, 2012. [Google Scholar]
- Benfenati, E. Theory, Guidance and Applications on QSAR and REACH; Orchestra: Milan, Italy, 2012; Available online: http://ebook.insilico.eu/insilico-ebook-orchestra-benfenati-ed1_rev-June2013.pdf (accessed on 24 November 2017).
- Roy, K. Advances in QSAR Modeling. Applications in Pharmaceutical, Chemical, Food, Agricultural and Environmental Sciences; Springer International Publishing: Berlin, Germany, 2017. [Google Scholar]
- Katritzky, A.R.; Goordeva, E.V. Traditional Topological Indices vs. Electronic, Geometrical, and Combined Molecular Descriptors in QSAR/QSPR Research. J. Chem. Inf. Comput. Sci. 1993, 33, 835–857. [Google Scholar] [CrossRef] [PubMed]
- Diudea, M.V.E. QSPR/QSAR Studies by Molecular Descriptors; Nova Science Publishers: New York, NY, USA, 2001. [Google Scholar]
- Todeschini, R.; Consonni, V. Molecular Descriptors for Chemoinformatics (Methods and Principles in Medicinal Chemistry); Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Masand, V.H.; Rastija, V. PyDescriptor: A new PyMOL plugin for calculating thousands of easily understandable molecular descriptors. Chemom. Intell. Lab. Syst. 2017, 169, 12–18. [Google Scholar] [CrossRef]
- Cao, S. QSAR, molecular docking studies of thiophene and imidazopyridine derivatives as polo-like kinase 1 inhibitors. J. Mol. Struct. 2012, 1020, 167–176. [Google Scholar] [CrossRef]
- Comelli, N.C.; Duchowicz, P.R.; Castro, E.A. QSAR models for thiophene and imidazopyridine derivatives inhibitors of the Polo-Like Kinase 1. Eur. J. Pharm. Sci. 2014, 62, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Chekkara, R.; Kandakatla, N.; Gorla, V.R.; Tenkayala, S.R.; Susithra, E. Theoretical studies on benzimidazole and imidazo [1, 2-a] pyridine derivatives as Polo-like kinase 1 (Plk1) inhibitors: Pharmacophore modeling, atom-based 3D-QSAR and molecular docking approach. J. Saudi Chem. Soc. 2017, 21, S311–S321. [Google Scholar] [CrossRef]
- Kong, Y.; Yan, A. QSAR models for predicting the bioactivity of Polo-Like Kinase 1 inhibitors. Chemom. Intell. Lab. Syst. 2017, 167, 214–225. [Google Scholar] [CrossRef]
- Duchowicz, P.R.; Comelli, N.C.; Ortiz, E.V.; Castro, E.A. QSAR study for carcinogenicity in a large set of organic compounds. Curr. Drug Saf. 2012, 7, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Duchowicz, P.R.; Bennardi, D.O.; Baselo, D.E.; Bonifazi, E.L.; Rios-Luci, C.; Padrón, J.M.; Burton, G.; Misico, R.I. QSAR on Antiproliferative Naphthoquinones Based on a Conformation-Independent Approach. Eur. J. Med. Chem. 2014, 77, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, E.V.; Bennardi, D.O.; Bacelo, D.E.; Fioressi, S.E.; Duchowicz, P.R. The conformation-independent QSPR approach for predicting the oxidation rate constant of water micropollutants. Environ. Sci. Pollut. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aranda, J.F.; Bacelo, D.E.; Leguizamón Aparicio, M.S.; Ocsachoque, M.A.; Castro, E.A.; Duchowicz, P.R. Predicting the Bioconcentration Factor through a Conformation-Independent QSPR Study. SAR&QSAR Environ. Res. 2017, 28, 749–763. [Google Scholar]
- Kothe, M.; Kohls, D.; Low, S.; Coli, R.; Cheng, A.C.; Jacques, S.L.; Johnson, T.L.; Lewis, C.; Loh, C.; Nonomiya, J.; et al. Structure of the catalytic domain of human polo-like kinase 1. Biochemistry 2007, 46, 5960–5971. [Google Scholar] [CrossRef] [PubMed]
- Kubinyi, H. (Ed.) Comparative Molecular Field Analysis (CoMFA); Wiley-VCH Verlag GmbH: Weinheim, Germany, 2003. [Google Scholar]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- ChEMBL. Available online: https://www.ebi.ac.uk/chembl/ (accessed on 24 November 2017).
- Open Babel for Windows. Available online: http://openbabel.org/wiki/Category:Installation (accessed on 24 November 2017).
- ACD/ChemSketch. Available online: www.acdlabs.com (accessed on 24 November 2017).
- Pharmaceutical Data Exploration Laboratory (PaDEL). Available online: http://www.yapcwsoft.com/ (accessed on 24 November 2017).
- Yap, C.W. PaDEL-Descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xie, Q.; Ge, W.; Qian, F.; Fang, H.; Shi, L.; Su, Z.; Perkins, R.; Tong, W. Mold2, molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Valdes-Martini, J.R.; García Jacas, C.R.; Marrero-Ponce, Y.; Silveira Vaz‘d Almeida, Y.; Morrel, C. QuBiLS-MAS: Free Software for Molecular Descriptors Calculator from Quadratic, Bilinear and Linear Maps Based on Graph–Theoretic Electronic-Density Matrices and Atomic Weightings; Version 1.0; CAMD-BIR Unit, CENDA Number of Register: 2373-2012; Central University of Las Villas: Villa Clara, Cuba, 2012. [Google Scholar]
- Duchowicz, P.R.; Castro, E.A.; Fernández, F.M. Alternative Algorithm for the Search of an Optimal Set of Descriptors in QSAR-QSPR Studies. MATCH Commun. Math. Comput. Chem. 2006, 55, 179–192. [Google Scholar]
- Morales, A.H.; Duchowicz, P.R.; Cabrera Pérez, M.A.; Castro, E.A.; Cordeiro, M.N.D.S.; González, M.P. Application of the replacement method as a novel variable selection strategy in QSAR. 1. Carcinogenic potential. Chemom. Intell. Lab. Syst. 2006, 81, 180–187. [Google Scholar] [CrossRef]
- Rojas, C.; Duchowicz, P.R.; Tripaldi, P.; Pis Diez, R. Quantitative structure-property relationship analysis for the retention index of fragrance-like compounds on a polar stationary phase. J. Chromatogr. A 2015, 1422, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Duchowicz, P.R.; Fioressi, S.E.; Castro, E.A.; Wróbel, K.; Ibezim, N.E.; Bacelo, D.E. Conformation-independent QSAR study on human epidermal growth factor receptor-2 (HER2) inhibitors. Chem. Sel. 2017, 2, 3725–3731. [Google Scholar] [CrossRef]
- Kaufman, L.; Rousseeuw, P.J. Finding Groups in Data: An Introduction to Cluster Analysis; Wiley: New York, NY, USA, 2005. [Google Scholar]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Gramatica, P. Principles of QSAR models validation: Internal and external. QSAR Comb. Sci. 2007, 26, 694–701. [Google Scholar] [CrossRef]
- Roy, K.; Kar, S.; Ambure, P. On a simple approach for determining applicability domain of QSAR models. Chemom. Intell. Lab. Syst. 2015, 145, 22–29. [Google Scholar] [CrossRef]
- Eriksson, L.; Jaworska, J.; Worth, A.P.; Cronin, M.T.; McDowell, R.M.; Gramatica, P. Methods for Reliability and Uncertainty Assessment and for Applicability Evaluations of Classification- and Regression-Based QSARs. Environ. Health Perspect. 2003, 111, 1361–1375. [Google Scholar] [CrossRef] [PubMed]
- Rücker, C.; Rücker, G.; Meringer, M. Y-Randomization and its variants in QSPR/QSAR. J. Chem. Inf. Model. 2007, 47, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Roy, P.P. Comparative chemometric modeling of cytochrome 3A4 inhibitory activity of structurally diverse compounds using stepwise MLR, FAMLR, PLS, GFA, G/PLS and ANN techniques. Eur. J. Med. Chem. 2009, 44, 2913–2922. [Google Scholar] [CrossRef] [PubMed]
- Gramatica, P.; Sangion, A. A Historical Excursus on the Statistical Validation Parameters for QSAR Models: A Clarification Concerning Metrics and Terminology. J. Chem. Inf. Model. 2016, 56, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Cooper, J.; Saracci, R.; Cole, P. Describing the validity of carcinogen screening tests. Br. J. Cancer 1979, 39, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.; Minkin, V.I.; Carpenter, B.K. Ockham’s Razor and Chemistry. Bull. Soc. Chim. Fr. 1996, 133, 117–130. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d | Descriptors | ||||||
|---|---|---|---|---|---|---|---|
| 1 | Sub99 | 0.31 | 1.18 | 0.39 | 1.25 | 0.28 | 1.31 |
| 2 | PC534; AP170 | 0.49 | 1.02 | 0.56 | 1.08 | 0.52 | 1.06 |
| 3 | PC534; KR4261; AP170 | 0.52 | 0.99 | 0.68 | 0.95 | 0.57 | 0.98 |
| 4 | nHBAcc3; PC534; KR4261; AP170 | 0.57 | 0.94 | 0.71 | 0.90 | 0.62 | 0.93 |
| 5 | PC534; KR3577; KR4268; AP170; KRC3897 | 0.61 | 0.90 | 0.71 | 0.89 | 0.71 | 0.83 |
| 6 | maxHCsats; M66; PC534; KR3577; KR4268; KRC3897 | 0.64 | 0.87 | 0.74 | 0.85 | 0.69 | 0.84 |
| 7 | maxHCsats; M66; PC534; PC686; KR3577; KR4268; AP159 | 0.66 | 0.84 | 0.74 | 0.84 | 0.66 | 0.89 |
| 8 | mindssC; maxHCsats; M66; PC494; PC534; PC686; KR3577; KR4268 | 0.69 | 0.80 | 0.75 | 0.82 | 0.69 | 0.85 |
| 9 | mindssC; maxHCsats; M66; PC494; PC534; PC686; KR3577; KR4268; APC510 | 0.70 | 0.79 | 0.75 | 0.82 | 0.70 | 0.85 |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duchowicz, P.R. Linear Regression QSAR Models for Polo-Like Kinase-1 Inhibitors. Cells 2018, 7, 13. https://doi.org/10.3390/cells7020013
Duchowicz PR. Linear Regression QSAR Models for Polo-Like Kinase-1 Inhibitors. Cells. 2018; 7(2):13. https://doi.org/10.3390/cells7020013
Chicago/Turabian StyleDuchowicz, Pablo R. 2018. "Linear Regression QSAR Models for Polo-Like Kinase-1 Inhibitors" Cells 7, no. 2: 13. https://doi.org/10.3390/cells7020013
APA StyleDuchowicz, P. R. (2018). Linear Regression QSAR Models for Polo-Like Kinase-1 Inhibitors. Cells, 7(2), 13. https://doi.org/10.3390/cells7020013