Unconventional Secretion and Intercellular Transfer of Mutant Huntingtin


Abstract
:1. Introduction
2. Unconventional Secretion and Intercellular Transport of Neuropathogenic Factors
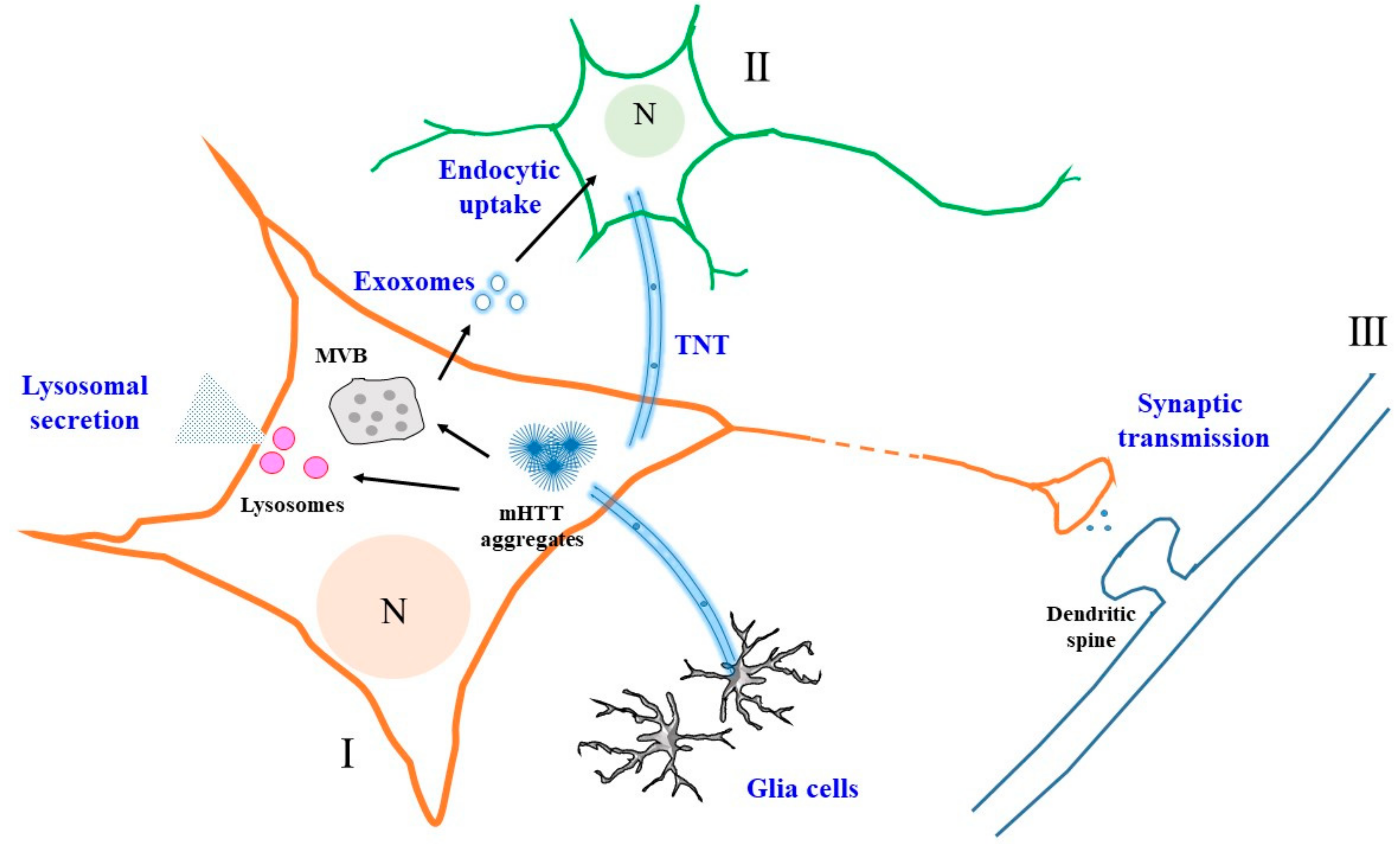
3. Externalization and Intercellular Transfer of mHTT
4. Mechanisms Underlying the Extracellular Secretion of mHTT—Exosomal, Autophagic or Lysosomal Secretion?
5. Prion-Like Spread of mHTT Aggregates?
6. Epilogue
Funding
Acknowledgments
Conflicts of Interest
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Den Dunnen, W.F.A. Trinucleotide repeat disorders. Handb. Clin. Neurol. 2017, 145, 383–391. [Google Scholar] [PubMed]
- Zeitlin, S.; Liu, J.P.; Chapman, D.L.; Papaioannou, V.E.; Efstratiadis, A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington’s disease gene homologue. Nat. Genet. 1995, 11, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef]
- Duyao, M.P.; Auerbach, A.B.; Ryan, A.; Persichetti, F.; Barnes, G.T.; McNeil, S.M.; Ge, P.; Vonsattel, J.P.; Gusella, J.F.; Joyner, A.L. Inactivation of the mouse Huntington’s disease gene homolog Hdh. Science 1995, 269, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.A.; Bork, P. HEAT repeats in the Huntington’s disease protein. Nat. Genet. 1995, 11, 115–116. [Google Scholar] [CrossRef] [PubMed]
- Seong, I.S.; Woda, J.M.; Song, J.J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.M.; Wheeler, V.C.; Walz, T.; et al. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA. 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudou, F.; Humbert, S. The biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.Y.; Zhou, X.F. Huntingtin associated protein 1 and its functions. Cell Adhes. Migr. 2009, 3, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, K.D.; Lim, Y.; Duffield, M.D.; Chataway, T.; Zhou, X.F.; Keating, D.J. Huntingtin-associated protein-1 (HAP1) regulates endocytosis and interacts with multiple trafficking-related proteins. Cell Signal. 2017, 35, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.P.; Banerjee, M.; Majumder, P. Huntington’s disease: Roles of huntingtin-interacting protein 1 (HIP-1) and its molecular partner HIPPI in the regulation of apoptosis and transcription. FEBS J. 2008, 275, 4271–4279. [Google Scholar] [CrossRef] [PubMed]
- McGuire, J.R.; Rong, J.; Li, S.H.; Li, X.J. Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J. Biol. Chem. 2006, 281, 3552–3559. [Google Scholar] [CrossRef] [PubMed]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L.F. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steffan, J.S.; Kazantsev, A.; Spasic-Boskovic, O.; Greenwald, M.; Zhu, Y.Z.; Gohler, H.; Wanker, E.E.; Bates, G.P.; Housman, D.E.; Thompson, L.M. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc. Natl. Acad. Sci. USA 2000, 97, 6763–6768. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Cheng, A.L.; Zhou, H.; Lam, S.; Rao, M.; Li, H.; Li, X.J. Interaction of Huntington disease protein with transcriptional activator Sp1. Mol. Cell. Biol. 2002, 22, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Tartari, M.; Crotti, A.; Goffredo, D.; Valenza, M.; Conti, L.; Cataudella, T.; Leavitt, B.R.; Hayden, M.R.; Timmusk, T.; et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003, 35, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Godin, J.D.; Colombo, K.; Molina-Calavita, M.; Keryer, G.; Zala, D.; Charrin, B.C.; Dietrich, P.; Volvert, M.L.; Guillemot, F.; Dragatsis, I.; et al. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron 2010, 67, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Keryer, G.; Pineda, J.R.; Liot, G.; Kim, J.; Dietrich, P.; Benstaali, C.; Smith, K.; Cordelières, F.P.; Spassky, N.; Ferrante, R.J.; et al. Ciliogenesis is regulated by a huntingtin-HAP1-PCM1 pathway and is altered in Huntington disease. J. Clin. Investig. 2011, 121, 4372–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haremaki, T.; Deglincerti, A.; Brivanlou, A.H. Huntingtin is required for ciliogenesis and neurogenesis during early Xenopus development. Dev. Biol. 2015, 408, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Karam, A.; Tebbe, L.; Weber, C.; Messaddeq, N.; Morlé, L.; Kessler, P.; Wolfrum, U.; Trottier, Y. A novel function of Huntingtin in the cilium and retinal ciliopathy in Huntington’s disease mice. Neurobiol. Dis. 2015, 80, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Holzbaur, E.L.F. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Ha, T.J.; Liu, L.; Nishimoto, A.; Reiner, A.; Goldowitz, D. Spatial and temporal requirements for huntingtin (Htt) in neuronal migration and survival during brain development. J. Neurosci. 2011, 31, 14794–14799. [Google Scholar] [CrossRef] [PubMed]
- Ben M’Barek, K.; Pla, P.; Orvoen, S.; Benstaali, C.; Godin, J.D.; Gardier, A.M.; Saudou, F.; David, D.J.; Humbert, S. Huntingtin mediates anxiety/depression-related behaviors and hippocampal neurogenesis. J. Neurosci. 2013, 33, 8608–8620. [Google Scholar] [CrossRef] [PubMed]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Sathasivam, K.; Neueder, A.; Gipson, T.A.; Landles, C.; Benjamin, A.C.; Bondulich, M.K.; Smith, D.L.; Faull, R.L.M.; Roos, R.A.C.; Howland, D.; et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, 2366–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Kim, M.; Roh, J.K.; Yoon, B.W.; Kang, L.; Kim, Y.J.; Aronin, N.; DiFiglia, M. Huntingtin is degraded to small fragments by calpain after ischemic injury. Exp. Neurol. 2003, 183, 109–115. [Google Scholar] [CrossRef]
- Gafni, J.; Hermel, E.; Young, J.E.; Wellington, C.L.; Hayden, M.R.; Ellerby, L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: Accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004, 279, 20211–20220. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Yi, Y.; Sapp, E.; Wang, Y.; Cuiffo, B.; Kegel, K.B.; Qin, Z.H.; Aronin, N.; DiFiglia, M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc. Natl. Acad. Sci. USA 2001, 98, 12784–12789. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.K.; Deng, Y.; Slow, E.J.; Haigh, B.; Bissada, N.; Lu, G.; Pearson, J.; Shehadeh, J.; Bertram, L.; Murphy, Z.; et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006, 125, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Warby, S.C.; Doty, C.N.; Graham, R.K.; Carroll, J.B.; Yang, Y.Z.; Singaraja, R.R.; Overall, C.M.; Hayden, M.R. Activated caspase-6 and caspase-6-cleaved fragments of huntingtin specifically colocalize in the nucleus. Hum. Mol. Genet. 2008, 17, 2390–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.J.; Sapp, E.; Cuiffo, B.G.; Sobin, L.; Yoder, J.; Kegel, K.B.; Qin, Z.H.; Detloff, P.; Aronin, N.; DiFiglia, M. Lysosomal proteases are involved in generation of N-terminal huntingtin fragments. Neurobiol. Dis. 2006, 22, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.P.; Holcomb, J.; Al-Ramahi, I.; de Haro, M.; Gafni, J.; Zhang, N.; Kim, E.; Sanhueza, M.; Torcassi, C.; Kwak, S.; et al. Matrix metalloproteinases are modifiers of huntingtin proteolysis and toxicity in Huntington’s disease. Neuron 2010, 67, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.A.; Jiang, H.; Ross, C.A. A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: Evidence for a compact beta-sheet structure. Hum. Mol. Genet. 2005, 14, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, P.; Pan, Y.; Sun, X.; Yang, H.; Difiglia, M.; Lu, B. A toxic mutant huntingtin species is resistant to selective autophagy. Nat. Chem. Biol. 2017, 13, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Gasset-Rosa, F.; Chillon-Marinas, C.; Goginashvili, A.; Atwal, R.S.; Artates, J.W.; Tabet, R.; Wheeler, V.C.; Bang, A.G.; Cleveland, D.W.; Lagier-Tourenne, C. Polyglutamine-expanded Huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron 2017, 94, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Lessmann, V.; Humbert, S.; et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Falk, J.D.; Vargiu, P.; Foye, P.E.; Usui, H.; Perez, J.; Danielson, P.E.; Lerner, D.L.; Bernal, J.; Sutcliffe, J.G. Rhes: A striatal-specific Ras homolog related to Dexras1. J. Neurosci. Res. 1999, 57, 782–788. [Google Scholar] [CrossRef]
- Subramaniam, S.; Sixt, K.M.; Barrow, R.; Snyder, S.H. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009, 324, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Swarnkar, S.; Chen, Y.; Pryor, W.M.; Shahani, N.; Page, D.T.; Subramaniam, S. Ectopic expression of the striatal-enriched GTPase Rhes elicits cerebellar degeneration and an ataxia phenotype in Huntington’s disease. Neurobiol. Dis. 2015, 82, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef] [PubMed]
- Zappulli, V.; Friis, K.P.; Fitzpatrick, Z.; Maguire, C.A.; Breakefield, X.O. Extracellular vesicles and intercellular communication within the nervous system. J. Clin. Investig. 2016, 126, 1198–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciregia, F.; Urbani, A.; Palmisano, G. Extracellular vesicles in brain tumors and neurodegenerative diseases. Front. Mol. Neurosci. 2017, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Victoria, G.S.; Zurzolo, C. The spread of prion-like proteins by lysosomes and tunneling nanotubes: Implications for neurodegenerative diseases. J. Cell Biol. 2017, 216, 2633–2644. [Google Scholar] [CrossRef] [PubMed]
- Pecho-Vrieseling, E.; Rieker, C.; Fuchs, S.; Bleckmann, D.; Esposito, M.S.; Botta, P.; Goldstein, C.; Bernhard, M.; Galimberti, I.; Müller, M.; et al. Transneuronal propagation of mutant huntingtin contributes to non-cell autonomous pathology in neurons. Nat. Neurosci. 2014, 17, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Babcock, D.T.; Ganetzky, B. Transcellular spreading of huntingtin aggregates in the Drosophila brain. Proc. Natl. Acad. Sci. USA 2015, 112, E5427–E5433. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Dai, W.; van Erp, T.G.M.; Overman, J.; Demuro, A.; Digman, M.A.; Hatami, A.; Albay, R.; Sontag, E.M.; Potkin, K.T.; et al. Huntington’s disease cerebrospinal fluid seeds aggregation of mutant huntingtin. Mol. Psychiatry 2015, 20, 1286–1293. [Google Scholar] [CrossRef] [PubMed]
- Jeon, I.; Cicchetti, F.; Cisbani, G.; Lee, S.; Li, E.; Bae, J.; Lee, N.; Li, L.; Im, W.; Kim, M.; et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 2016, 132, 577–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trajkovic, K.; Jeong, H.; Krainc, D. Mutant huntingtin is secreted via a late endosomal/lysosomal unconventional secretory pathway. J. Neurosci. 2017, 37, 9000–9012. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Abounit, S.; Marzo, L.; Danckaert, A.; Chamoun, Z.; Roux, P.; Zurzolo, C. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 2013, 126, 3678–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, M.M.P.; Spartz, E.J.; Hong, W.; Luo, L.; Kopito, R.R. Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun. 2015, 6, 6768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, N.K.; Isas, J.M.; Rawat, A.; Lee, R.V.; Langen, J.; Pandey, P.; Langen, R. The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 2018, 293, 2597–2605. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Tabrizi, S.J. The ubiquitin-proteasome system in neurodegeneration. Antioxid. Redox Signal. 2014, 21, 2302–2321. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, D.J.; García-Arencibia, M.; Hochfeld, W.E.; Rubinsztein, D.C. Autophagy and misfolded proteins in neurodegeneration. Exp. Neurol. 2012, 238, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.E.L.; Lim, Y.S.; Lee, M.G.; Tang, B.L. Non-classical membrane trafficking processes galore. J. Cell. Physiol. 2012, 227, 3722–3730. [Google Scholar] [CrossRef] [PubMed]
- Rabouille, C.; Malhotra, V.; Nickel, W. Diversity in unconventional protein secretion. J. Cell Sci. 2012, 125, 5251–5255. [Google Scholar] [CrossRef] [PubMed]
- Ng, F.; Tang, B.L. Unconventional Protein Secretion in Animal Cells. Methods Mol. Biol. 2016, 1459, 31–46. [Google Scholar] [PubMed]
- Cruz-Garcia, D.; Malhotra, V.; Curwin, A.J. Unconventional protein secretion triggered by nutrient starvation. Semin. Cell Dev. Biol. 2018, S1084-9521(18)30003-X. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Sedgwick, A.E.; D’Souza-Schorey, C. The biology of extracellular microvesicles. Traffic 2018, 19, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Fuxe, K. Extracellular-vesicle type of volume transmission and tunnelling-nanotube type of wiring transmission add a new dimension to brain neuro-glial networks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130505. [Google Scholar] [CrossRef] [PubMed]
- Janas, A.M.; Sapoń, K.; Janas, T.; Stowell, M.H.B.; Janas, T. Exosomes and other extracellular vesicles in neural cells and neurodegenerative diseases. Biochim. Biophys. Acta 2016, 1858, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Levy, E. Exosomes in the diseased brain: First insights from in vivo studies. Front. Neurosci. 2017, 11, 142. [Google Scholar] [CrossRef] [PubMed]
- Cocucci, E.; Meldolesi, J. Ectosomes and exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barr, M.M. Ciliary Extracellular Vesicles: Txt Msg Organelles. Cell. Mol. Neurobiol. 2016, 36, 449–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quezada, C.; Torres, Á.; Niechi, I.; Uribe, D.; Contreras-Duarte, S.; Toledo, F.; San Martín, R.; Gutiérrez, J.; Sobrevia, L. Role of extracellular vesicles in glioma progression. Mol. Aspects Med. 2018, 60, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; Wani, M.R.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar] [PubMed]
- Jiang, D.; Gao, F.; Zhang, Y.; Wong, D.S.H.; Li, Q.; Tse, H.F.; Xu, G.; Yu, Z.; Lian, Q. Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 2016, 7, e2467. [Google Scholar] [CrossRef] [PubMed]
- Naphade, S.; Sharma, J.; Gaide Chevronnay, H.P.; Shook, M.A.; Yeagy, B.A.; Rocca, C.J.; Ur, S.N.; Lau, A.J.; Courtoy, P.J.; Cherqui, S. Lysosomal cross-correction by hematopoietic stem cell-derived macrophages via tunneling nanotubes. Stem Cells 2015, 33, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar α-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. Cell biology and pathophysiology of α-Synuclein. Cold Spring Harb. Perspect. Med. 2018, 8, a024091. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yu, S. Synucleinopathies: Common features and hippocampal manifestations. Cell. Mol. Life Sci. 2017, 74, 1485–1501. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef] [PubMed]
- Jang, A.; Lee, H.J.; Suk, J.E.; Jung, J.W.; Kim, K.P.; Lee, S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274. [Google Scholar] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances α-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Clairembault, T.; Biraud, M.; Neunlist, M.; Derkinderen, P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J. Neurochem. 2013, 125, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Minakaki, G.; Keramioti, M.V.; Xylaki, M.; Balafas, E.; Chrysanthou-Piterou, M.; Kloukina, I.; Vekrellis, K. GABA transmission via ATP-dependent K+ channels regulates α-synuclein secretion in mouse striatum. Brain 2016, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Cho, E.D.; Lee, K.W.; Kim, J.H.; Cho, S.G.; Lee, S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013, 45, e22. [Google Scholar] [CrossRef] [PubMed]
- Poehler, A.M.; Xiang, W.; Spitzer, P.; May, V.E.L.; Meixner, H.; Rockenstein, E.; Chutna, O.; Outeiro, T.F.; Winkler, J.; Masliah, E.; et al. Autophagy modulates SNCA/α-synuclein release, thereby generating a hostile microenvironment. Autophagy 2014, 10, 2171–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Qin, M.; Bao, P.; Xu, W.; Xu, J. Secretory carrier membrane protein 5 is an autophagy inhibitor that promotes the secretion of α-synuclein via exosome. PLoS ONE 2017, 12, e0180892. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Ejlerskov, P.; Rasmussen, I.; Vilhardt, F. Reciprocal signals between microglia and neurons regulate α-synuclein secretion by exophagy through a neuronal cJUN-N-terminal kinase-signaling axis. J. Neuroinflamm. 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Eitan, E.; Wu, T.Y.; Mattson, M.P. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging 2018, 61, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.J.; Kim, D.; Park, J.; Zhao, H.; Lee, S.J.; Chang, S. The secreted oligomeric form of α-synuclein affects multiple steps of membrane trafficking. FEBS Lett. 2013, 587, 452–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieri, G.; Gitler, A.D.; Brahic, M. Internalization, axonal transport and release of fibrillar forms of alpha-synuclein. Neurobiol. Dis. 2018, 109, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A.; Ximerakis, M.; Vekrellis, K. Protein transmission, seeding and degradation: Key steps for α-Synuclein prion-like propagation. Exp. Meurol. 2014, 23, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Rostami, J.; Holmqvist, S.; Lindström, V.; Sigvardson, J.; Westermark, G.T.; Ingelsson, M.; Bergström, J.; Roybon, L.; Erlandsson, A. Human astrocytes transfer aggregated alpha-Synuclein via tunneling nanotubes. J. Neurosci. 2017, 37, 11835–11853. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Simón, D.; García-García, E.; Gómez-Ramos, A.; Falcón-Pérez, J.M.; Díaz-Hernández, M.; Hernández, F.; Avila, J. Tau overexpression results in its secretion via membrane vesicles. Neurodegener. Dis. 2012, 10, 73–75. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Dage, J.L.; Citron, M. Constitutive secretion of tau protein by an unconventional mechanism. Neurobiol. Dis. 2012, 48, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Kanmert, D.; Cantlon, A.; Muratore, C.R.; Jin, M.; O’Malley, T.T.; Lee, G.; Young-Pearse, T.L.; Selkoe, D.J.; Walsh, D.M. C-terminally truncated Forms of Tau, but not full-Length Tau or its C-terminal fragments, are released from neurons independently of cell death. J. Neurosci. 2015, 35, 10851–10865. [Google Scholar] [CrossRef] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, L.; Leissring, M.A. Insulin-degrading enzyme is exported via an unconventional protein secretion pathway. Mol. Neurodegener. 2009, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Cha, M.Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.S.; Park, S.A.; Mook-Jung, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamboli, I.Y.; Barth, E.; Christian, L.; Siepmann, M.; Kumar, S.; Singh, S.; Tolksdorf, K.; Heneka, M.T.; Lütjohann, D.; Wunderlich, P.; et al. Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J. Biol. Chem. 2010, 285, 37405–37414. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Kang, S.; Choi, H.; Mook-Jung, I. Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol. Neurodegener. 2015, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.J.; Atkin, J.D.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.F.; Cheema, S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu, Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, D.; Brouwers, N.; Duran, J.M.; Mora, G.; Curwin, A.J.; Malhotra, V. A diacidic motif determines unconventional secretion of wild-type and ALS-linked mutant SOD1. J. Cell Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iguchi, Y.; Eid, L.; Parent, M.; Soucy, G.; Bareil, C.; Riku, Y.; Kawai, K.; Takagi, S.; Yoshida, M.; Katsuno, M.; et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain 2016, 139, 3187–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamelgarn, M.; Chen, J.; Kuang, L.; Arenas, A.; Zhai, J.; Zhu, H.; Gal, J. Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochim. Biophys. Acta 2016, 1862, 2004–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feiler, M.S.; Strobel, B.; Freischmidt, A.; Helferich, A.M.; Kappel, J.; Brewer, B.M.; Li, D.; Thal, D.R.; Walther, P.; Ludolph, A.C.; et al. TDP-43 is intercellularly transmitted across axon terminals. J. Cell Biol. 2015, 211, 897–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Ma, M.; Teng, J.; Teng, R.K.F.; Zhou, S.; Yin, J.; Fonkem, E.; Huang, J.H.; Wu, E.; Wang, X. Exposure to ALS-FTD-CSF generates TDP-43 aggregates in glioblastoma cells through exosomes and TNTs-like structure. Oncotarget 2015, 6, 24178–24191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fevrier, B.; Vilette, D.; Archer, F.; Loew, D.; Faigle, W.; Vidal, M.; Laude, H.; Raposo, G. Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. USA 2004, 101, 9683–9688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Vilette, D.; Courte, J.; Peyrin, J.M.; Coudert, L.; Schaeffer, L.; Andréoletti, O.; Leblanc, P. Cellular mechanisms responsible for cell-to-cell spreading of prions. Cell. Mol. Life Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.H.; Lauckner, J.E.; Kachirskaia, I.; Heuser, J.E.; Melki, R.; Kopito, R.R. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 2009, 11, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wild, E.J.; Boggio, R.; Langbehn, D.; Robertson, N.; Haider, S.; Miller, J.R.C.; Zetterberg, H.; Leavitt, B.R.; Kuhn, R.; Tabrizi, S.J.; et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J. Clin. Investig. 2015, 125, 1979–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Abels, E.R.; Redzic, J.S.; Margulis, J.; Finkbeiner, S.; Breakefield, X.O. Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington’s disease: Background and evaluation in cell culture. Cell. Mol. Neurobiol. 2016, 36, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Cicchetti, F.; Lacroix, S.; Cisbani, G.; Vallières, N.; Saint-Pierre, M.; St-Amour, I.; Tolouei, R.; Skepper, J.N.; Hauser, R.A.; Mantovani, D.; et al. Mutant huntingtin is present in neuronal grafts in Huntington disease patients. Ann. Neurol. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.L. Identification of the major steps in botulinum toxin action. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Hidalgo, L.; Altuntas, S.; Rossin, F.; D’Eletto, M.; Marsella, C.; Farrace, M.G.; Falasca, L.; Antonioli, M.; Fimia, G.M.; Piacentini, M. Transglutaminase type 2-dependent selective recruitment of proteins into exosomes under stressful cellular conditions. Biochim. Biophys. Acta 2016, 1863, 2084–2092. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant huntingtin inhibits αB-crystallin expression and impairs exosome secretion from astrocytes. J. Neurosci. 2017, 37, 9550–9563. [Google Scholar] [CrossRef] [PubMed]
- Claude-Taupin, A.; Jia, J.; Mudd, M.; Deretic, V. Autophagy’s secret life: Secretion instead of degradation. Essays Biochem. 2017, 61, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Gee, H.Y.; Noh, S.H.; Tang, B.L.; Kim, K.H.; Lee, M.G. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell 2011, 146, 746–760. [Google Scholar] [CrossRef] [PubMed]
- Duran, J.M.; Anjard, C.; Stefan, C.; Loomis, W.F.; Malhotra, V. Unconventional secretion of Acb1 is mediated by autophagosomes. J. Cell Biol. 2010, 188, 527–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjithaya, R.; Anjard, C.; Loomis, W.F.; Subramani, S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J. Cell Biol. 2010, 188, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjithaya, R.; Subramani, S. Role of autophagy in unconventional protein secretion. Autophagy 2010, 6, 650–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heng, M.Y.; Duong, D.K.; Albin, R.L.; Tallaksen-Greene, S.J.; Hunter, J.M.; Lesort, M.J.; Osmand, A.; Paulson, H.L.; Detloff, P.J. Early autophagic response in a novel knock-in model of Huntington disease. Hum. Mol. Genet. 2010, 19, 3702–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, J.; Bassaganyas, L.; Lepreux, S.; Chiritoiu, M.; Costet, P.; Ripoche, J.; Malhotra, V.; Schekman, R. Unconventional secretion of FABP4 by endosomes and secretory lysosomes. J. Cell Biol. 2018, 217, 649–665. [Google Scholar] [CrossRef] [PubMed]
- van der Sluijs, P.; Zibouche, M.; van Kerkhof, P. Late steps in secretory lysosome exocytosis in cytotoxic lymphocytes. Front. Immunol. 2013, 4, 359. [Google Scholar] [CrossRef] [PubMed]
- Andrews, N.W. Regulated secretion of conventional lysosomes. Trends Cell Biol. 2000, 10, 316–321. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Giasson, B.I.; Borchelt, D.R. Prion-like spreading in tauopathies. Biol. Psychiatry 2018, 83, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Hasegawa, M. TDP-43 Prions. Cold Spring Harb. Perspect. Med. 2018, 8, a024463. [Google Scholar] [CrossRef] [PubMed]
- Kim, M. Beta conformation of polyglutamine track revealed by a crystal structure of Huntingtin N-terminal region with insertion of three histidine residues. Prion 2013, 7, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, A.; Muth, C.; Dabrowski, O.; Krasemann, S.; Glatzel, M. Exosomes and the Prion protein: More than one truth. Front. Neurosci. 2017, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Mudher, A.; Colin, M.; Dujardin, S.; Medina, M.; Dewachter, I.; Alavi Naini, S.M.; Mandelkow, E.M.; Mandelkow, E.; Buée, L.; Goedert, M.; et al. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol. Commun. 2017, 5, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Tu, Z.; Liu, Z.; Fan, N.; Yang, H.; Yang, S.; Yang, W.; Zhao, Y.; Ouyang, Z.; Lai, C.; et al. A Huntingtin knockin pig model recapitulates features of selective neurodegeneration in Huntington’s disease. Cell 2018, 173, 989–1002. [Google Scholar] [CrossRef] [PubMed]
- Leeman, D.S.; Hebestreit, K.; Ruetz, T.; Webb, A.E.; McKay, A.; Pollina, E.A.; Dulken, B.W.; Zhao, X.; Yeo, R.W.; Ho, T.T.; et al. Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 2018, 359, 1277–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Neurodegenerative Diseases | Unconventionally Secreted Neurotoxic Agent | Known Secretion/Intercellular Transfer Mechanisms | References |
|---|---|---|---|
| Alzheimer’s disease | Aβ peptides | Exosomes | [98] |
| Tau | EVs (including exosomes) | [100,101] | |
| TNT | [50,105] | ||
| Parkinson’s disease | α-Synuclein | Exosomes | [84,85] |
| TNT | [79,97] | ||
| Amyotrophic lateral sclerosis | SOD1 | Exosomes | [113] |
| TDP-43 | Exosomes | [114] | |
| TNT | [117] | ||
| FUS | Exosomes | [115] | |
| Huntington’s disease | mHTT | EVs (including exosomes) | [55,121] |
| TNT | [57] | ||
| Prion disease | PrPSc | Exosomes | [117] |
| TNT | [118] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.L. Unconventional Secretion and Intercellular Transfer of Mutant Huntingtin. Cells 2018, 7, 59. https://doi.org/10.3390/cells7060059
Tang BL. Unconventional Secretion and Intercellular Transfer of Mutant Huntingtin. Cells. 2018; 7(6):59. https://doi.org/10.3390/cells7060059
Chicago/Turabian StyleTang, Bor Luen. 2018. "Unconventional Secretion and Intercellular Transfer of Mutant Huntingtin" Cells 7, no. 6: 59. https://doi.org/10.3390/cells7060059
APA StyleTang, B. L. (2018). Unconventional Secretion and Intercellular Transfer of Mutant Huntingtin. Cells, 7(6), 59. https://doi.org/10.3390/cells7060059