The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
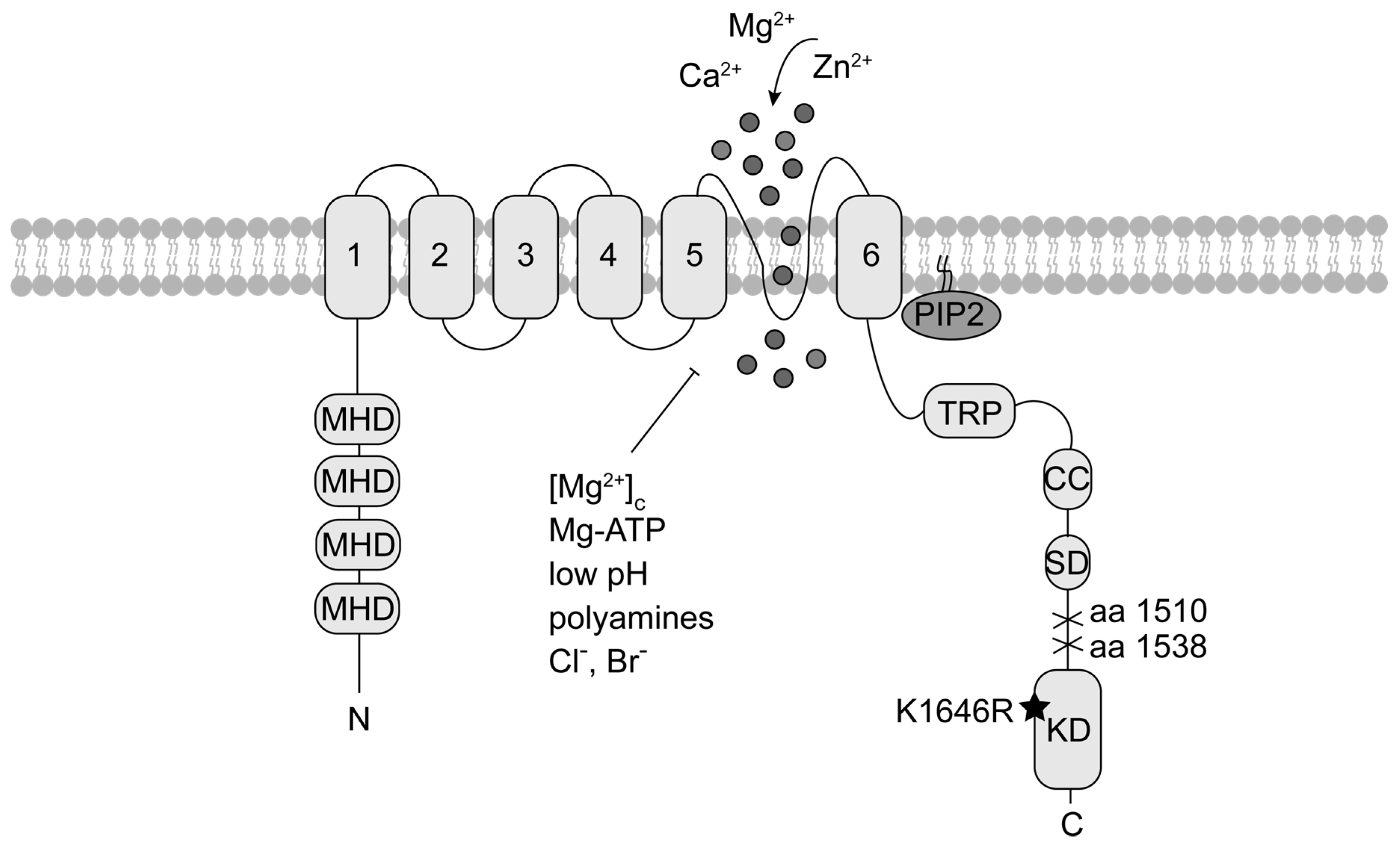
2. The Channel-Kinase TRPM7 in Immune Cell Signalling
2.1. TRPM7 Kinase Regulates Mast Cell Reactivity
2.2. TRPM7 in Neutrophil Migration
2.3. TRPM7 Guides Macrophage Activation and Polarization
2.4. TRPM7 Affects Lymphocyte Functions
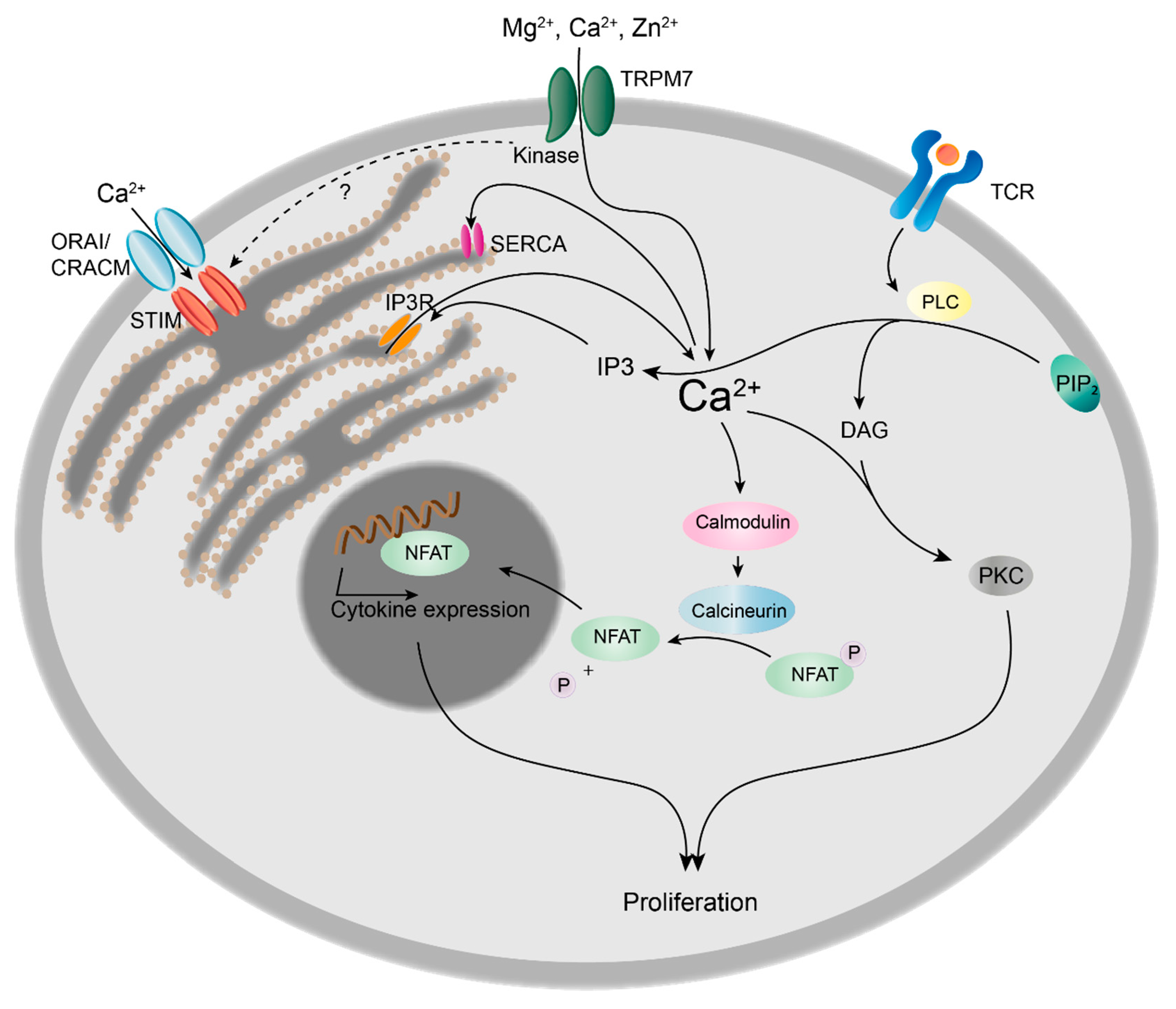
2.4.1. TRPM7 Kinase Regulates Intracellular Calcium Signals and Proliferation in Lymphocytes
2.4.2. TRPM7 in Cell Growth, Activation and Development of B Cells
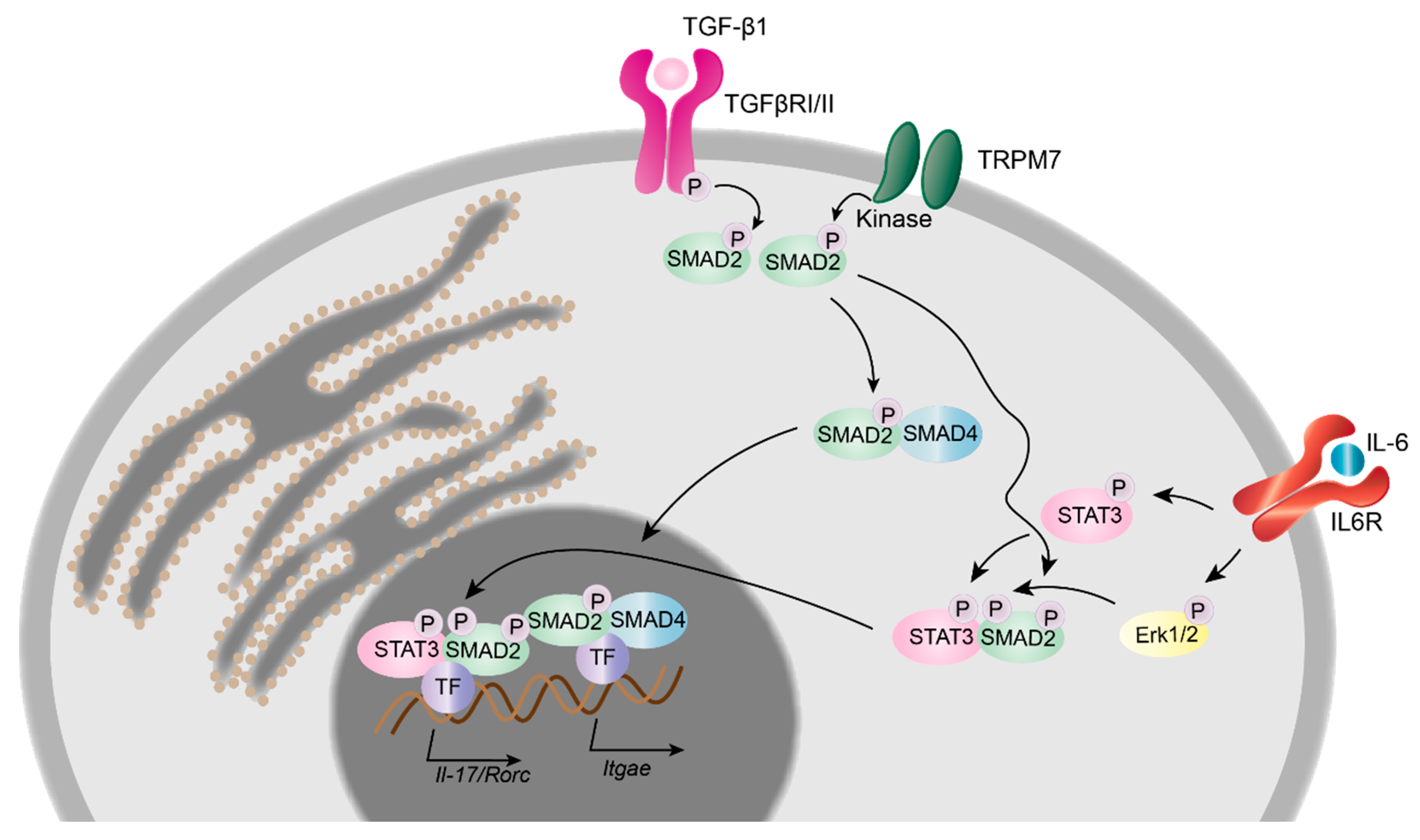
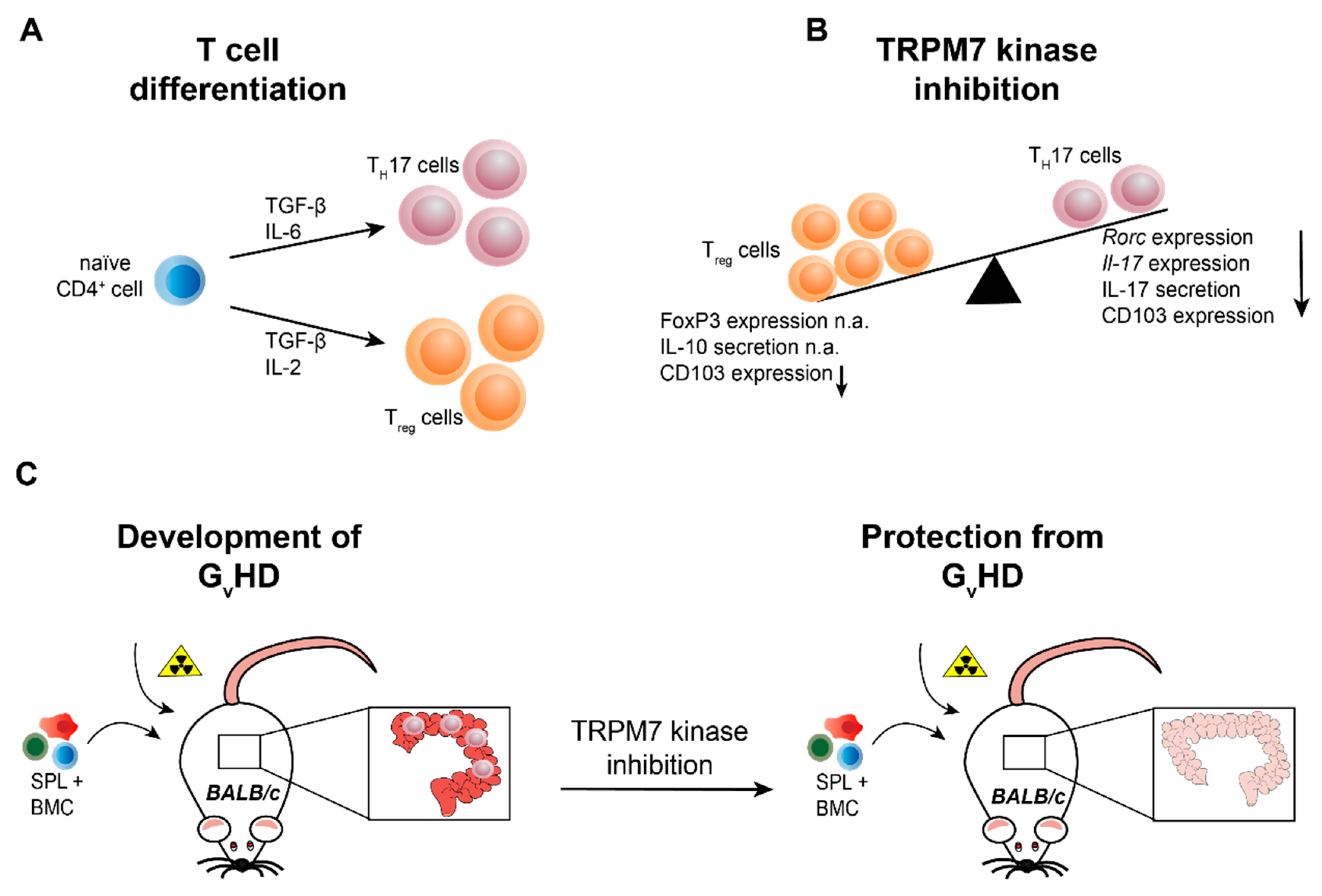
2.4.3. TRPM7 in Murine T Cell Development, Differentiation and Transcriptional Regulation
3. TRPM7-Mediated Hematologic and Inflammatory Diseases
3.1. Hypomagnesaemia and TRPM7 Kinase in Delayed-Type Hypersensitivity Reactions
3.2. The TRPM7 Channel-Kinase in Arterial Thrombosis and Stroke
3.3. TRPM7 Kinase Signalling Supports Graft versus Host Reactions
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Runnels, L.W.; Yue, L.; Clapham, D.E. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science 2001, 291, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Ryazanova, L.V.; Rondon, L.J.; Zierler, S.; Hu, Z.; Galli, J.; Yamaguchi, T.P.; Mazur, A.; Fleig, A.; Ryazanov, A.G. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 2010, 1, 109. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, C.; Perraud, A.L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef]
- He, Y.; Yao, G.; Savoia, C.; Touyz, R.M. Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells: Role of angiotensin II. Circ. Res. 2005, 96, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Abiria, S.A.; Krapivinsky, G.; Sah, R.; Santa-Cruz, A.G.; Chaudhuri, D.; Zhang, J.; Adstamongkonkul, P.; DeCaen, P.G.; Clapham, D.E. TRPM7 senses oxidative stress to release Zn2+ from unique intracellular vesicles. Proc. Natl. Acad. Sci. USA 2017, 114, E6079–E6088. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Branigan, D.; Xiong, Z.G. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J. Biol. Chem. 2010, 285, 7430–7439. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Lis, A.; Schmitz, C.; Penner, R.; Fleig, A. The TRPM7 kinase limits receptor-induced calcium release by regulating heterotrimeric G-proteins. Cell. Mol. Life Sci. 2018, 75, 3069–3078. [Google Scholar] [CrossRef] [PubMed]
- Beesetty, P.; Wieczerzak, K.B.; Gibson, J.N.; Kaitsuka, T.; Luu, C.T.; Matsushita, M.; Kozak, J.A. Inactivation of TRPM7 kinase in mice results in enlarged spleens, reduced T-cell proliferation and diminished store-operated calcium entry. Sci. Rep. 2018, 8, 3023. [Google Scholar] [CrossRef] [PubMed]
- Gotru, S.K.; Chen, W.; Kraft, P.; Becker, I.C.; Wolf, K.; Stritt, S.; Zierler, S.; Hermanns, H.M.; Rao, D.; Perraud, A.L.; et al. TRPM7 Kinase Controls Calcium Responses in Arterial Thrombosis and Stroke in Mice. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [PubMed]
- Demeuse, P.; Penner, R.; Fleig, A. TRPM7 channel is regulated by magnesium nucleotides via its kinase domain. J. Gen. Physiol. 2006, 127, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Kozak, J.A.; Matsushita, M.; Nairn, A.C.; Cahalan, M.D. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J. Gen. Physiol. 2005, 126, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Runnels, L.W.; Yue, L.; Clapham, D.E. The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat. Cell Biol. 2002, 4, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Zhang, Z.; Lis, A.; Penner, R.; Fleig, A. TRPM7 is regulated by halides through its kinase domain. Cell. Mol. Life Sci. 2013, 70, 2757–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romani, A.M. Cellular magnesium homeostasis. Arch. Biochem. Biophys. 2011, 512, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chubanov, V.; Ferioli, S.; Gudermann, T. Assessment of TRPM7 functions by drug-like small molecules. Cell Calcium 2017, 67, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Ferioli, S.; Zierler, S.; Zaisserer, J.; Schredelseker, J.; Gudermann, T.; Chubanov, V. TRPM6 and TRPM7 differentially contribute to the relief of heteromeric TRPM6/7 channels from inhibition by cytosolic Mg2+ and Mg.ATP. Sci. Rep. 2017, 7, 8806. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, A.; Vettore, V.; Rezzonico-Jost, T.; Hampe, S.; Rottoli, E.; Nadolni, W.; Perotti, M.; Meier, M.A.; Hermanns, C.; Geiger, S.; et al. TRPM7 kinase activity is essential for T cell colonization and alloreactivity in the gut. Nat. Commun. 2017, 8, 1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Matsushita, M.; Nairn, A.C.; Kuriyan, J. Crystal structure of the atypical protein kinase domain of a TRP channel with phosphotransferase activity. Mol. Cell 2001, 7, 1047–1057. [Google Scholar] [CrossRef]
- Ryazanova, L.V.; Dorovkov, M.V.; Ansari, A.; Ryazanov, A.G. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J. Biol. Chem. 2004, 279, 3708–3716. [Google Scholar] [CrossRef] [PubMed]
- Dorovkov, M.V.; Ryazanov, A.G. Phosphorylation of annexin I by TRPM7 channel-kinase. J. Biol. Chem. 2004, 279, 50643–50646. [Google Scholar] [CrossRef] [PubMed]
- Dorovkov, M.V.; Kostyukova, A.S.; Ryazanov, A.G. Phosphorylation of annexin A1 by TRPM7 kinase: A switch regulating the induction of an alpha-helix. Biochemistry 2011, 50, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Middelbeek, J.; Dorovkov, M.V.; Figdor, C.G.; Ryazanov, A.G.; Lasonder, E.; van Leeuwen, F.N. The alpha-kinases TRPM6 and TRPM7, but not eEF-2 kinase, phosphorylate the assembly domain of myosin IIA, IIB and IIC. FEBS Lett. 2008, 582, 2993–2997. [Google Scholar] [CrossRef] [PubMed]
- Deason-Towne, F.; Perraud, A.L.; Schmitz, C. Identification of Ser/Thr phosphorylation sites in the C2-domain of phospholipase C γ2 (PLCγ2) using TRPM7-kinase. Cell Signal. 2012, 24, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamoorthy, M.; Wasim, L.; Buhari, F.H.M.; Zhao, T.; Mahtani, T.; Ho, J.; Kang, S.; Deason-Towne, F.; Perraud, A.L.; Schmitz, C.; et al. The channel-kinase TRPM7 regulates antigen gathering and internalization in B cells. Sci. Signal. 2018, 11, eaah6692. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science 2008, 322, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Kaitsuka, T.; Katagiri, C.; Beesetty, P.; Nakamura, K.; Hourani, S.; Tomizawa, K.; Kozak, J.A.; Matsushita, M. Inactivation of TRPM7 kinase activity does not impair its channel function in mice. Sci. Rep. 2014, 4, 5718. [Google Scholar] [CrossRef] [PubMed]
- Ryazanova, L.V.; Hu, Z.; Suzuki, S.; Chubanov, V.; Fleig, A.; Ryazanov, A.G. Elucidating the role of the TRPM7 alpha-kinase: TRPM7 kinase inactivation leads to magnesium deprivation resistance phenotype in mice. Sci. Rep. 2014, 4, 7599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, B.N.; Krapivinsky, G.; Navarro, B.; Krapivinsky, L.; Carter, B.C.; Febvay, S.; Delling, M.; Penumaka, A.; Ramsey, I.S.; Manasian, Y.; et al. Cleavage of TRPM7 releases the kinase domain from the ion channel and regulates its participation in Fas-induced apoptosis. Dev. Cell 2012, 22, 1149–1162. [Google Scholar] [CrossRef] [PubMed]
- Zierler, S.; Sumoza-Toledo, A.; Suzuki, S.; Duill, F.O.; Ryazanova, L.V.; Penner, R.; Ryazanov, A.G.; Fleig, A. TRPM7 kinase activity regulates murine mast cell degranulation. J. Physiol. 2016, 594, 2957–2970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Shin, S.K.; Song, M.Y.; Lee, J.E.; Park, K.S. Identification of the phosphorylation sites on intact TRPM7 channels from mammalian cells. Biochem. Biophys. Res. Commun. 2012, 417, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.; Middelbeek, J.; Morrice, N.A.; Figdor, C.G.; Lasonder, E.; van Leeuwen, F.N. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS ONE 2008, 3, e1876. [Google Scholar] [CrossRef] [PubMed]
- Brandao, K.; Deason-Towne, F.; Zhao, X.; Perraud, A.L.; Schmitz, C. TRPM6 kinase activity regulates TRPM7 trafficking and inhibits cellular growth under hypomagnesic conditions. Cell. Mol. Life Sci. 2014, 71, 4853–4867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, N.; Bai, Z.; Nanda, V.; Runnels, L.W. Mass Spectrometric Analysis of TRPM6 and TRPM7 Phosphorylation Reveals Regulatory Mechanisms of the Channel-Kinases. Sci. Rep. 2017, 7, 42739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonfria, E.; Murdock, P.R.; Cusdin, F.S.; Benham, C.D.; Kelsell, R.E.; McNulty, S. Tissue distribution profiles of the human TRPM cation channel family. J. Recept. Signal Transduct. 2006, 26, 159–178. [Google Scholar] [CrossRef] [PubMed]
- Zierler, S.; Hampe, S.; Nadolni, W. TRPM channels as potential therapeutic targets against pro-inflammatory diseases. Cell Calcium 2017, 67, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Schappe, M.S.; Szteyn, K.; Stremska, M.E.; Mendu, S.K.; Downs, T.K.; Seegren, P.V.; Mahoney, M.A.; Dixit, S.; Krupa, J.K.; Stipes, E.J.; et al. Chanzyme TRPM7 Mediates the Ca2+ Influx Essential for Lipopolysaccharide-Induced Toll-Like Receptor 4 Endocytosis and Macrophage Activation. Immunity 2018, 48, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Siebenhaar, F.; Redegeld, F.A.; Bischoff, S.C.; Gibbs, B.F.; Maurer, M. Mast Cells as Drivers of Disease and Therapeutic Targets. Trends Immunol. 2017, 39, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Selvakumar, G.P.; Thangavel, R.; Ahmed, M.E.; Zaheer, S.; Raikwar, S.P.; Iyer, S.S.; Bhagavan, S.M.; Beladakere-Ramaswamy, S.; Zaheer, A. Mast Cell Activation in Brain Injury, Stress, and Post-traumatic Stress Disorder and Alzheimer’s Disease Pathogenesis. Front. Neurosci. 2017, 11, 703. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. Neuroinflammation, Mast Cells, and Glia: Dangerous Liaisons. Neuroscientist 2017, 23, 478–498. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, K.A.; Kovanen, P.T. Isolation of mast cell granules. Curr. Protoc. Cell Biol. 2006, 29, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Bulfone-Paus, S.; Nilsson, G.; Draber, P.; Blank, U.; Levi-Schaffer, F. Positive and Negative Signals in Mast Cell Activation. Trends Immunol. 2017, 38, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.P.; Thomson, D.S. Histamine release from rat mast cells passively sensitised with homocytotropic (IgE) antibody. Int. Arch. Allergy Appl. Immunol. 1972, 43, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chang, Y.C.; Chang, H.A.; Lin, Y.S.; Tsao, C.W.; Shen, M.R.; Chiu, W.T. Differential Ca2+ mobilization and mast cell degranulation by FcepsilonRI- and GPCR-mediated signaling. Cell Calcium 2017, 67, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Gaudenzio, N.; Sibilano, R.; Marichal, T.; Starkl, P.; Reber, L.L.; Cenac, N.; McNeil, B.D.; Dong, X.; Hernandez, J.D.; Sagi-Eisenberg, R.; et al. Different activation signals induce distinct mast cell degranulation strategies. J. Clin. Investig. 2016, 126, 3981–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Nishida, K.; Fujii, Y.; Hirano, T.; Hikida, M.; Kurosaki, T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat. Immunol. 2008, 9, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Vig, M.; DeHaven, W.I.; Bird, G.S.; Billingsley, J.M.; Wang, H.; Rao, P.E.; Hutchings, A.B.; Jouvin, M.H.; Putney, J.W.; Kinet, J.P. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 2008, 9, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Wykes, R.C.; Lee, M.; Duffy, S.M.; Yang, W.; Seward, E.P.; Bradding, P. Functional transient receptor potential melastatin 7 channels are critical for human mast cell survival. J. Immunol. 2007, 179, 4045–4052. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Ng, N.M.; Chen, M.; Lin, X.; Tang, T.; Cheng, H.; Yang, C.; Jiang, S. Inhibition of TRPM7 channels reduces degranulation and release of cytokines in rat bone marrow-derived mast cells. Int. J. Mol. Sci. 2014, 15, 11817–11831. [Google Scholar] [CrossRef] [PubMed]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [PubMed]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Simon, S.I. Chemokines, selectins and intracellular calcium flux: Temporal and spatial cues for leukocyte arrest. Front. Immunol. 2012, 3, 188. [Google Scholar] [CrossRef] [PubMed]
- Immler, R.; Simon, S.I.; Sperandio, M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Investig. 2018, e12964. [Google Scholar] [CrossRef] [PubMed]
- Abed, E.; Moreau, R. Importance of melastatin-like transient receptor potential 7 and magnesium in the stimulation of osteoblast proliferation and migration by PDGF. Am. J. Physiol. Cell Physiol. 2009, 297, C360–C368. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.P.; Luan, Y.; You, C.X.; Chen, X.H.; Luo, R.C.; Li, R. TRPM7 regulates the migration of human nasopharyngeal carcinoma cell by mediating Ca2+ influx. Cell Calcium 2010, 47, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Rong, M.Y.; Wang, L.; Ren, Z.; Chen, L.N.; Jia, J.F.; Li, X.Y.; Wu, Z.B.; Chen, Z.N.; Zhu, P. CD147 up-regulates calcium-induced chemotaxis, adhesion ability and invasiveness of human neutrophils via a TRPM-7-mediated mechanism. Rheumatology 2014, 53, 2288–2296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.W.; Liu, H.; Li, X.D.; Sui, S.G.; Liu, Y.F. Salvianolic acid B protects against acute lung injury by decreasing TRPM6 and TRPM7 expressions in a rat model of sepsis. J. Cell. Biochem. 2018, 119, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef] [PubMed]
- Santoni, G.; Morelli, M.B.; Amantini, C.; Santoni, M.; Nabissi, M.; Marinelli, O.; Santoni, A. “Immuno-Transient Receptor Potential Ion Channels”: The Role in Monocyte- and Macrophage-Mediated Inflammatory Responses. Front. Immunol. 2018, 9, 1273. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Im, Y.J.; Kim, Y.L.; Im, D.S. Characterization of Ca2+ influx induced by dimethylphytosphingosine and lysophosphatidylcholine in U937 monocytes. Biochem. Biophys. Res. Commun. 2006, 348, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Wuensch, T.; Thilo, F.; Krueger, K.; Scholze, A.; Ristow, M.; Tepel, M. High glucose-induced oxidative stress increases transient receptor potential channel expression in human monocytes. Diabetes 2010, 59, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jiang, H.; Ruan, C.; Zhong, J.; Gao, P.; Zhu, D.; Niu, W.; Guo, S. The interaction of transient receptor potential melastatin 7 with macrophages promotes vascular adventitial remodeling in transverse aortic constriction rats. Hypertens. Res. 2014, 37, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.; Miralles, F.; Eder, C. TRPM7 regulates proliferation and polarisation of macrophages. J. Cell Sci. 2014, 127, 4561–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Chubanov, V.; Mederos y Schnitzler, M.; Meissner, M.; Schafer, S.; Abstiens, K.; Hofmann, T.; Gudermann, T. Natural and synthetic modulators of SK (Kca2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. Br. J. Pharmacol. 2012, 166, 1357–1376. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Zee, I.; Concepcion, A.R.; Maus, M.; Shaw, P.; Portal-Celhay, C.; Zahra, A.; Kozhaya, L.; Weidinger, C.; Philips, J.; et al. Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells. J. Immunol. 2015, 195, 1202–1217. [Google Scholar] [CrossRef] [PubMed]
- Haslberger, A.; Romanin, C.; Koerber, R. Membrane potential modulates release of tumor necrosis factor in lipopolysaccharide-stimulated mouse macrophages. Mol. Biol. Cell 1992, 3, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Yatim, K.M.; Lakkis, F.G. A brief journey through the immune system. Clin. J. Am. Soc. Nephrol. 2015, 10, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, S.; Marchingo, J.M.; Horton, M.B.; Hodgkin, P.D. The regulation of lymphocyte activation and proliferation. Curr. Opin. Immunol. 2018, 51, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Kozak, J.A.; Shimizu, Y.; McLachlin, D.T.; Yamaguchi, H.; Wei, F.Y.; Tomizawa, K.; Matsui, H.; Chait, B.T.; Cahalan, M.D.; et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J. Biol. Chem. 2005, 280, 20793–20803. [Google Scholar] [CrossRef] [PubMed]
- Sahni, J.; Scharenberg, A.M. TRPM7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 2008, 8, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Sahni, J.; Tamura, R.; Sweet, I.R.; Scharenberg, A.M. TRPM7 regulates quiescent/proliferative metabolic transitions in lymphocytes. Cell Cycle 2010, 9, 3565–3574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat. Commun. 2016, 7, 11097. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, M.; Buhari, F.H.M.; Zhao, T.; Brauer, P.M.; Burrows, K.; Cao, E.Y.; Moxley-Paquette, V.; Mortha, A.; Zuniga-Pflucker, J.C.; Treanor, B. The ion channel TRPM7 is required for B cell lymphopoiesis. Sci. Signal. 2018, 11, eaan2693. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, F.; Cheroutre, H. Mucosal T cells in gut homeostasis and inflammation. Expert Rev. Clin. Immunol. 2010, 6, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamada, N.; Nunez, G. Role of the gut microbiota in the development and function of lymphoid cells. J. Immunol. 2013, 190, 1389–1395. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Butcher, E.C. Rapid acquisition of tissue-specific homing phenotypes by CD4+ T cells activated in cutaneous or mucosal lymphoid tissues. J. Exp. Med. 2002, 195, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Wang, W.; Xiao, W.; Yang, H. Role of the intestinal cytokine microenvironment in shaping the intraepithelial lymphocyte repertoire. J. Leukoc. Biol. 2015, 97, 849–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Habtezion, A.; Nguyen, L.P.; Hadeiba, H.; Butcher, E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology 2016, 150, 340–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, M.P.; Arya, A.; Murphy, E.A.; Adams, C.M.; Strauch, U.G.; Agace, W.W.; Marsal, J.; Donohue, J.P.; Her, H.; Beier, D.R.; et al. Mucosal T lymphocyte numbers are selectively reduced in integrin alpha E (CD103)-deficient mice. J. Immunol. 1999, 162, 6641–6649. [Google Scholar] [PubMed]
- Mokrani, M.; Klibi, J.; Bluteau, D.; Bismuth, G.; Mami-Chouaib, F. Smad and NFAT pathways cooperate to induce CD103 expression in human CD8 T lymphocytes. J. Immunol. 2014, 192, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Porzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar] [CrossRef] [PubMed]
- Kuras, Z.; Yun, Y.H.; Chimote, A.A.; Neumeier, L.; Conforti, L. KCa3.1 and TRPM7 channels at the uropod regulate migration of activated human T cells. PLoS ONE 2012, 7, e43859. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.; Hilewitz, A. Hypomagnesaemia in patients hospitalised in internal medicine is associated with increased mortality. Int. J. Clin. Pract. 2014, 68, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Trapani, V.; Wolf, F.I.; Scaldaferri, F. Dietary magnesium: The magic mineral that protects from colon cancer? Magnes. Res. 2015, 28, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Chubanov, V.; Ferioli, S.; Wisnowsky, A.; Simmons, D.G.; Leitzinger, C.; Einer, C.; Jonas, W.; Shymkiv, Y.; Bartsch, H.; Braun, A.; et al. Epithelial magnesium transport by TRPM6 is essential for prenatal development and adult survival. eLife 2016, 5, e20914. [Google Scholar] [CrossRef] [PubMed]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nature 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpuech-Brugere, C.; Nowacki, W.; Daveau, M.; Gueux, E.; Linard, C.; Rock, E.; Lebreton, J.; Mazur, A.; Rayssiguier, Y. Inflammatory response following acute magnesium deficiency in the rat. Biochim. Biophys. Acta 2000, 1501, 91–98. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Reddy, P. Pathophysiology of graft-versus-host disease. Semin. Hematol. 2006, 43, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Filipovich, A.H.; Weisdorf, D.; Pavletic, S.; Socie, G.; Wingard, J.R.; Lee, S.J.; Martin, P.; Chien, J.; Przepiorka, D.; Couriel, D.; et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol. Blood Marrow Transplant. 2005, 11, 945–956. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, J.L.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadolni, W.; Zierler, S. The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis. Cells 2018, 7, 109. https://doi.org/10.3390/cells7080109
Nadolni W, Zierler S. The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis. Cells. 2018; 7(8):109. https://doi.org/10.3390/cells7080109
Chicago/Turabian StyleNadolni, Wiebke, and Susanna Zierler. 2018. "The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis" Cells 7, no. 8: 109. https://doi.org/10.3390/cells7080109
APA StyleNadolni, W., & Zierler, S. (2018). The Channel-Kinase TRPM7 as Novel Regulator of Immune System Homeostasis. Cells, 7(8), 109. https://doi.org/10.3390/cells7080109