Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications



Abstract
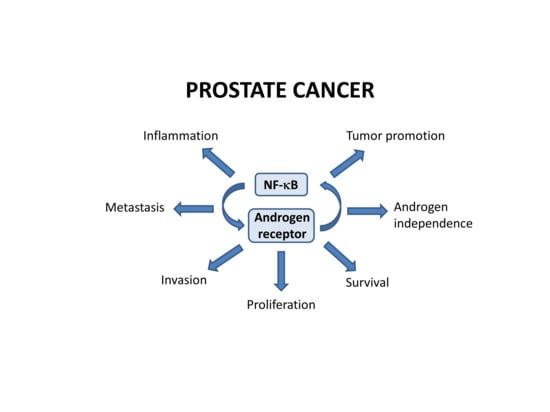
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. The NF-κB Family of Transcription Factors
1.2. Prostate Cancer: Role of Inflammation and Androgen Hormones
2. NF-κB in Prostate Cancer
2.1. NF-κB in Prostate Cancer Cell Survival and Proliferation
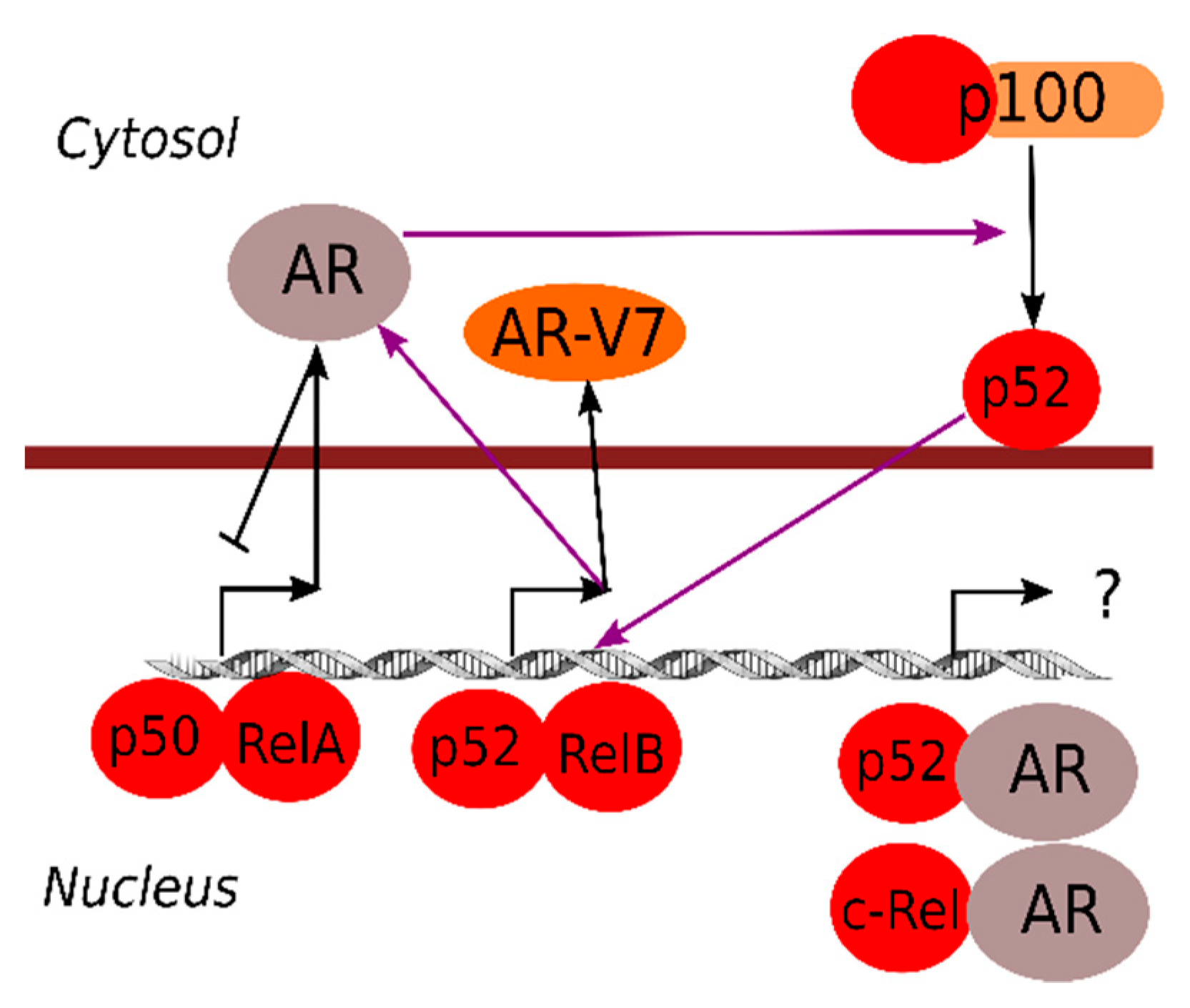
2.2. Crosstalk between NF-κB and Androgen Receptor Signaling
3. NF-κB Signaling Pathways in Prostate Cancers
3.1. Innate Immune Receptors in Prostate Cancer
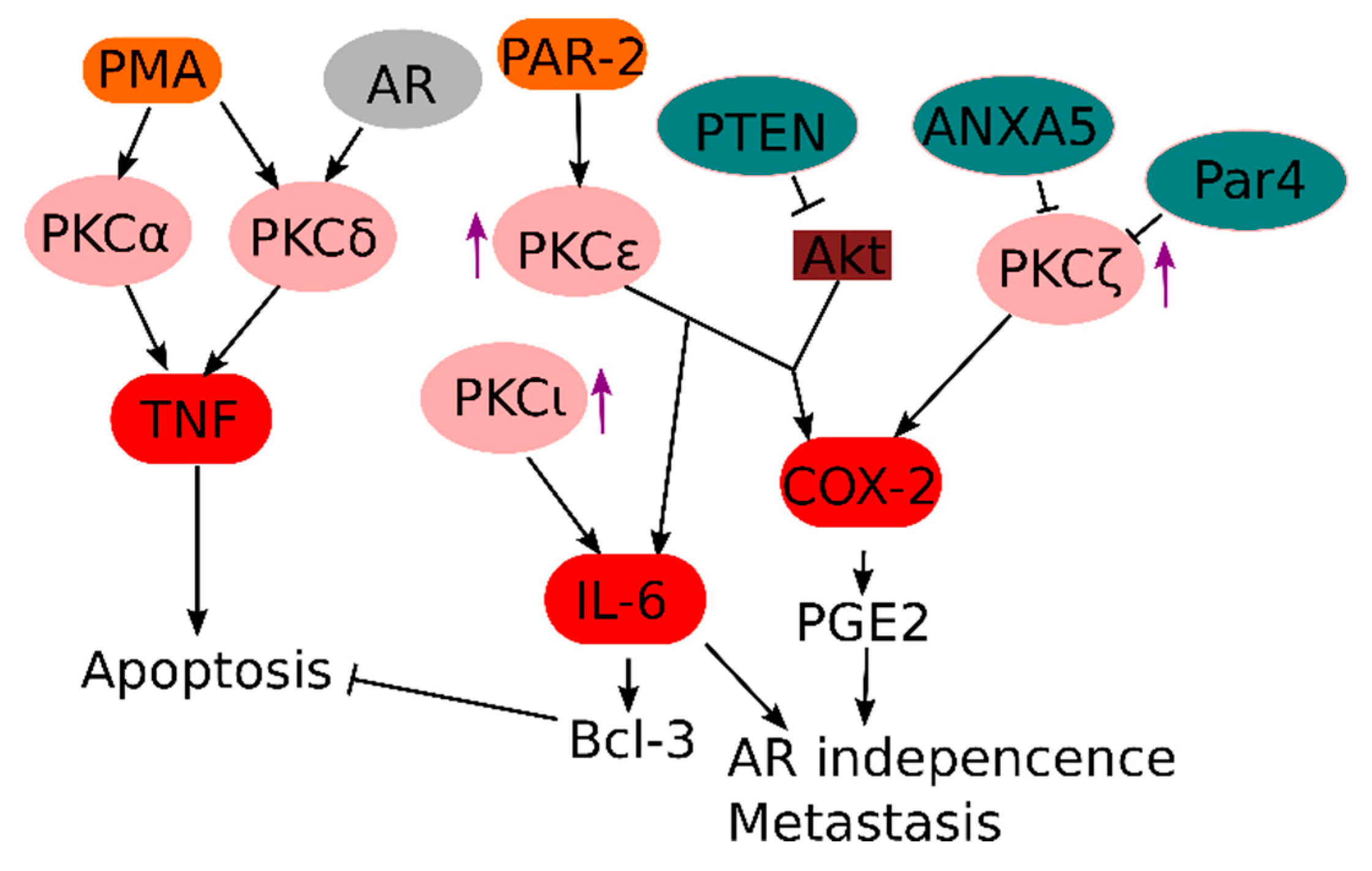
3.2. PKC and PKC-Related Signals in Prostate Cancer
3.3. GPCR Signaling in Prostate Cancer
4. NF-κB-Induced Risk Factors in Prostate Cancer
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Medicina. Available online: https://www.wdl.org/en/item/11618/ (accessed on 28 August 2018).
- Virchow, R. Die krankhaften Geschwülste: Strumen, Myome, Neurome, Angiome; Verlag Von August Hirschwald: Berlin, Germany, 1863. [Google Scholar]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonina, I.S.; Zhong, Z.; Karin, M.; Beyaert, R. Limiting inflammation-the negative regulation of NF-κB and the NLRP3 inflammasome. Nat. Immunol. 2017, 18, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Kochel, T.; Rice, N.R. v-rel- and c-rel-protein complexes bind to the NF-kappa B site in vitro. Oncogene 1992, 7, 567–572. [Google Scholar] [PubMed]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef] [PubMed]
- Clément, J.-F.; Meloche, S.; Servant, M.J. The IKK-related kinases: From innate immunity to oncogenesis. Cell Res. 2008, 18, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Wietek, C.; Cleaver, C.S.; Ludbrook, V.; Wilde, J.; White, J.; Bell, D.J.; Lee, M.; Dickson, M.; Ray, K.P.; O’Neill, L.A.J. IkappaB kinase epsilon interacts with p52 and promotes transactivation via p65. J. Biol. Chem. 2006, 281, 34973–34981. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Leslie, J.; Perkins, N.D. c-Rel and its many roles in cancer: An old story with new twists. Br. J. Cancer 2016, 114, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, T.D.; Gerondakis, S. The c-Rel Transcription Factor in Development and Disease. Genes Cancer 2011, 2, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.-E.; Guzzardi, M.; Décaillet, C.; Grau, M.; Dörken, B.; et al. Malt1-Dependent RelB Cleavage Promotes Canonical NF-κB Activation in Lymphocytes and Lymphoma Cell Lines. Proc. Natl. Acad. Sci. USA 2011, 108, 14596–14601. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Aleksiyadis, K.; Martin, A.; McNamee, K.; Tharmalingam, T.; Williams, R.O.; Mémet, S.; Cope, A.P. Inhibitor of kappa B epsilon (IκBε) is a non-redundant regulator of c-Rel-dependent gene expression in murine T and B cells. PLoS ONE 2011, 6, e24504. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.; Hayden, M.S.; Long, M.; Scott, M.L.; West, A.P.; Zhang, D.; Oeckinghaus, A.; Lynch, C.; Hoffmann, A.; Baltimore, D.; et al. IkappaBbeta acts to inhibit and activate gene expression during the inflammatory response. Nature 2010, 466, 1115–1119. [Google Scholar] [CrossRef] [PubMed]
- McKenna, S.; Wright, C.J. Inhibiting IκBβ-NFκB signaling attenuates the expression of select pro-inflammatory genes. J. Cell Sci. 2015, 128, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Tsui, R.; Kearns, J.D.; Lynch, C.; Vu, D.; Ngo, K.A.; Basak, S.; Ghosh, G.; Hoffmann, A. IκBβ enhances the generation of the low-affinity NFκB/RelA homodimer. Nat. Commun. 2015, 6, 7068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, E.; Hildebrand, D.G.; Kriebs, A.; Obermayer, K.; Manz, M.; Rothfuss, O.; Schulze-Osthoff, K.; Essmann, F. IκBζ is a regulator of the senescence-associated secretory phenotype in DNA damage- and oncogene-induced senescence. J. Cell Sci. 2013, 126, 3738–3745. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Chmielarska, K.; Hennecke, K.; Pfeifer, A.; Fässler, R. Cyld Inhibits Tumor Cell Proliferation by Blocking Bcl-3-Dependent NF-κB Signaling. Cell 2006, 125, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, V.; Melendez-Zajgla, J. Role of Bcl-3 in solid tumors. Mol. Cancer 2011, 10, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, G.P.; Delongchamps, N.; Brawley, O.W.; Wang, C.Y.; de la Roza, G. The Worldwide Epidemiology of Prostate Cancer: Perspectives from Autopsy Studies. Can. J. Urol. 2008, 15, 3866–3871. [Google Scholar] [PubMed]
- Biancardi, M.F.; dos Santos, F.C.A.; de Carvalho, H.F.; Sanches, B.D.A.; Taboga, S.R. Female prostate: Historical, developmental, and morphological perspectives. Cell Biol. Int. 2017, 41, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Risberg, G.; Johansson, E.E.; Hamberg, K. A theoretical model for analysing gender bias in medicine. Int. J. Equity Health 2009, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pongtippan, A.; Malpica, A.; Levenback, C.; Deavers, M.T.; Silva, E.G. Skene’s gland adenocarcinoma resembling prostatic adenocarcinoma. Int. J. Gynecol. Pathol. 2004, 23, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Leroy, B.E.; Northrup, N. Prostate cancer in dogs: Comparative and clinical aspects. Vet. J. Lond. Engl. 1997 2009, 180, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.K.; Elshafae, S.M.; Keller, E.T.; McCauley, L.K.; Rosol, T.J. Review of Animal Models of Prostate Cancer Bone Metastasis. Vet. Sci. 2014, 1, 16–39. [Google Scholar] [CrossRef]
- Di Sebastiano, K.M.; Pinthus, J.H.; Duivenvoorden, W.C.M.; Mourtzakis, M. Glucose impairments and insulin resistance in prostate cancer: The role of obesity, nutrition and exercise. Obes. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Caffo, O.; Galli, L.; Maugeri, A.; Scarpi, E.; Maines, F.; Chiuri, V.E.; Lolli, C.; Kinspergher, S.; Schepisi, G.; et al. Association among metabolic syndrome, inflammation, and survival in prostate cancer. Urol. Oncol. 2018, 36, 240.e1–240.e11. [Google Scholar] [CrossRef] [PubMed]
- Shankar, E.; Vykhovanets, E.V.; Vykhovanets, O.V.; Maclennan, G.T.; Singh, R.; Bhaskaran, N.; Shukla, S.; Gupta, S. High-fat diet activates pro-inflammatory response in the prostate through association of Stat-3 and NF-κB. Prostate 2012, 72, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.; Liou, G.-Y. Macrophage Cytokines Enhance Cell Proliferation of Normal Prostate Epithelial Cells through Activation of ERK and Akt. Sci. Rep. 2018, 8, 7718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ianni, M.; Porcellini, E.; Carbone, I.; Potenzoni, M.; Pieri, A.M.; Pastizzaro, C.D.; Benecchi, L.; Licastro, F. Genetic factors regulating inflammation and DNA methylation associated with prostate cancer. Prostate Cancer Prostatic Dis. 2013, 16, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Jerde, T.J.; Bushman, W. IL-1 induces IGF-dependent epithelial proliferation in prostate development and reactive hyperplasia. Sci. Signal. 2009, 2, ra49. [Google Scholar] [CrossRef] [PubMed]
- Penney, K.L.; Stampfer, M.J.; Jahn, J.L.; Sinnott, J.A.; Flavin, R.; Rider, J.R.; Finn, S.; Giovannucci, E.; Sesso, H.D.; Loda, M.; et al. Gleason Grade Progression Is Uncommon. Cancer Res. 2013, 73, 5163–5168. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bubendorf, L.; Schöpfer, A.; Wagner, U.; Sauter, G.; Moch, H.; Willi, N.; Gasser, T.C.; Mihatsch, M.J. Metastatic patterns of prostate cancer: An autopsy study of 1,589 patients. Hum. Pathol. 2000, 31, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Shou, J.; Zhang, Q.; Wang, S.; Zhang, D. The prognosis of different distant metastases pattern in prostate cancer: A population based retrospective study. Prostate 2018, 78, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Mokarram, P.; Alizadeh, J.; Razban, V.; Barazeh, M.; Solomon, C.; Kavousipour, S. Interconnection of Estrogen/Testosterone Metabolism and Mevalonate Pathway in Breast and Prostate Cancers. Curr. Mol. Pharmacol. 2017, 10, 86–114. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Davis, I.D.; Birrell, S.N.; Tilley, W.D. Breast and prostate cancer: More similar than different. Nat. Rev. Cancer 2010, 10, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C. Endocrine-induced regression of cancers. Cancer Res. 1967, 27, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen Levels Increase by Intratumoral De novo Steroidogenesis during Progression of Castration-Resistant Prostate Cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, J.A.F.; Bruni-Cardoso, A.; Augusto, T.M.; Damas-Souza, D.M.; Barbosa, G.O.; Felisbino, S.L.; Stach-Machado, D.R.; Carvalho, H.F. Macrophage roles in the clearance of apoptotic cells and control of inflammation in the prostate gland after castration. Prostate 2018, 78, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Mitri, D.D.; Gil, V.; Crespo, M.; Bernardis, G.D.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 1. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.C.; Strand, D.W.; Love, H.L.; Franco, O.E.; Grabowska, M.M.; Miller, N.L.; Hameed, O.; Clark, P.E.; Matusik, R.J.; Jin, R.J.; et al. NF-κB and androgen receptor variant 7 induce expression of SRD5A isoforms and confer 5ARI resistance. Prostate 2016, 76, 1004–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turo, R.; Jallad, S.; Prescott, S.; Cross, W.R. Metastatic prostate cancer in transsexual diagnosed after three decades of estrogen therapy. Can. Urol. Assoc. J. 2013, 7, E544–E546. [Google Scholar] [CrossRef] [PubMed]
- Miksad, R.A.; Bubley, G.; Church, P.; Sanda, M.; Rofsky, N.; Kaplan, I.; Cooper, A. Prostate Cancer in a Transgender Woman 41 Years After Initiation of Feminization. JAMA 2006, 296, 2312–2317. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.C.A.; Leite, R.P.; Custódio, A.M.G.; Carvalho, K.P.; Monteiro-Leal, L.H.; Santos, A.B.; Góes, R.M.; Carvalho, H.F.; Taboga, S.R. Testosterone Stimulates Growth and Secretory Activity of the Female Prostate in the Adult Gerbil (Meriones unguiculatus). Biol. Reprod. 2006, 75, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Obiezu, C.V.; Giltay, E.J.; Magklara, A.; Scorilas, A.; Gooren, L.J.; Yu, H.; Howarth, D.J.; Diamandis, E.P. Serum and urinary prostate-specific antigen and urinary human glandular kallikrein concentrations are significantly increased after testosterone administration in female-to-male transsexuals. Clin. Chem. 2000, 46, 859–862. [Google Scholar] [PubMed]
- Aultman, B. Cisgender. TSQ Transgender Stud. Q. 2014, 1, 61–62. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kregel, S.; Chen, J.L.; Tom, W.; Krishnan, V.; Kach, J.; Brechka, H.; Fessenden, T.B.; Isikbay, M.; Paner, G.P.; Szmulewitz, R.Z.; et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget 2016, 7, 26259–26274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colditz, J.; Rupf, B.; Maiwald, C.; Baniahmad, A. Androgens induce a distinct response of epithelial-mesenchymal transition factors in human prostate cancer cells. Mol. Cell. Biochem. 2016, 421, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, C.; Liu, F.; Li, H.-Z.; Peng, G.; Gao, X.; Dong, K.-Q.; Wang, H.-R.; Kong, D.-P.; Qu, M.; et al. Reciprocal Network between Cancer Stem-Like Cells and Macrophages Facilitates the Progression and Androgen Deprivation Therapy Resistance of Prostate Cancer. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Debelec-Butuner, B.; Alapinar, C.; Varisli, L.; Erbaykent-Tepedelen, B.; Hamid, S.M.; Gonen-Korkmaz, C.; Korkmaz, K.S. Inflammation-mediated abrogation of androgen signaling: An in vitro model of prostate cell inflammation. Mol. Carcinog. 2014, 53, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Ammirante, M.; Luo, J.-L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature 2010, 464, 302–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.P.; Bray, T.M.; Ho, E. Induction of proinflammatory response in prostate cancer epithelial cells by activated macrophages. Cancer Lett. 2009, 276, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Gray, K.P.; Harshman, L.C.; Evan, C.; Nakabayashi, M.; Fichorova, R.; Rider, J.; Mucci, L.; Kantoff, P.W.; Sweeney, C.J. Elevated IL-8, TNF-α, and MCP-1 in men with metastatic prostate cancer starting androgen-deprivation therapy (ADT) are associated with shorter time to castration-resistance and overall survival. Prostate 2014, 74, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten null prostate epithelium promotes localized myeloid-derived suppressor cell expansion and immune suppression during tumor initiation and progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Verzella, D.; Fischietti, M.; Capece, D.; Vecchiotti, D.; Del Vecchio, F.; Cicciarelli, G.; Mastroiaco, V.; Tessitore, A.; Alesse, E.; Zazzeroni, F. Targeting the NF-κB pathway in prostate cancer: A promising therapeutic approach? Curr. Drug Targets 2016, 17, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.; Perez, C.J.; Wang, H.; Benavides, F.J.; Kazanietz, M.G. Activation of nuclear factor κB (NF-κB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon). J. Biol. Chem. 2012, 287, 37570–37582. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Uzquiza, A.; Lopez-Haber, C.; Jernigan, D.L.; Fatatis, A.; Kazanietz, M.G. PKCε Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol. Cancer Res. 2015, 13, 1336–1346. [Google Scholar] [CrossRef] [PubMed]
- Longoni, N.; Sarti, M.; Albino, D.; Civenni, G.; Malek, A.; Ortelli, E.; Pinton, S.; Mello-Grand, M.; Ostano, P.; D’Ambrosio, G.; et al. ETS transcription factor ESE1/ELF3 orchestrates a positive feedback loop that constitutively activates NF-κB and drives prostate cancer progression. Cancer Res. 2013, 73, 4533–4547. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Lou, W.; Sun, M.; Chen, J.; Yue, J.; Kung, H.-J.; Evans, C.P.; Zhou, Q.; Gao, A.C. Aberrant activation of the androgen receptor by NF-kappaB2/p52 in prostate cancer cells. Cancer Res. 2010, 70, 3309–3319. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-H.; Park, S.-J.; Dickinson, S.I.; Luo, J.-L. A Constitutive Intrinsic Inflammatory Signaling Circuit Composed of miR-196b, Meis2, PPP3CC, and p65 Drives Prostate Cancer Castration Resistance. Mol. Cell 2017, 65, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yi, S.; Zhou, J.; Zhang, Y.; Guo, F. The NF-κB subunit RelB regulates the migration and invasion abilities and the radio-sensitivity of prostate cancer cells. Int. J. Oncol. 2016, 49, 381–392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.-C.; Qiu, T.; Dan, C.; Liu, X.-H.; Hu, C.-H. Blockage of RelB expression by gene silencing enhances the radiosensitivity of androgen-independent prostate cancer cells. Mol. Med. Rep. 2015, 11, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Caino, M.C.; Kazanietz, M.G. Regulation of Transcriptional Networks by PKC Isozymes: Identification of c-Rel as a Key Transcription Factor for PKC-Regulated Genes. PLoS ONE 2013, 8, e67319. [Google Scholar] [CrossRef] [PubMed]
- Ahlqvist, K.; Saamarthy, K.; Syed Khaja, A.S.; Bjartell, A.; Massoumi, R. Expression of Id proteins is regulated by the Bcl-3 proto-oncogene in prostate cancer. Oncogene 2013, 32, 1601–1608. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med. 2011, 17, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-initiating stem-like cells in human prostate cancer exhibit increased NF-κB signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, J.J.; Pandey, S.K.; Yadav, A.; Goel, S.; Ateeq, B. Targeting NF-kappa B Signaling by Artesunate Restores Sensitivity of Castrate-Resistant Prostate Cancer Cells to Antiandrogens. Neoplasia 2017, 19, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yamashita, H.; Yu, X.; Wang, J.; Franco, O.E.; Wang, Y.; Hayward, S.W.; Matusik, R.J. Inhibition of NF-kappa B signaling restores responsiveness of castrate-resistant prostate cancer cells to anti-androgen treatment by decreasing androgen receptor-variant expression. Oncogene 2015, 34, 3700–3710. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.J.; Lho, Y.; Connelly, L.; Wang, Y.; Yu, X.; Saint Jean, L.; Case, T.C.; Ellwood-Yen, K.; Sawyers, C.L.; Bhowmick, N.A.; et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008, 68, 6762–6769. [Google Scholar] [CrossRef] [PubMed]
- Viljoen, T.C.; van Aswegen, C.H.; du Plessis, D.J. Influence of acetylsalicylic acid and metabolites on DU-145 prostatic cancer cell proliferation. Oncology 1995, 52, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhou, W.; Ge, J.; Zhang, Z. Prostaglandin E2 receptor EP4 is involved in the cell growth and invasion of prostate cancer via the cAMP-PKA/PI3K-Akt signaling pathway. Mol. Med. Rep. 2018, 17, 4702–4712. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lee, Y.-F.; Li, G.; Liu, S.; Bao, B.-Y.; Huang, J.; Hsu, C.-L.; Chang, C. A new prostate cancer therapeutic approach: Combination of androgen ablation with COX-2 inhibitor. Int. J. Cancer 2008, 123, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.-J.; Lan, S.-W.; Lu, Y.-C.; Cheng, T.-S.; Lai, P.-F.; Tsai, C.-H.; Hsu, T.-W.; Lin, H.-Y.; Shyu, H.-Y.; Wu, S.-R.; et al. Inhibition of cyclooxygenase-2-mediated matriptase activation contributes to the suppression of prostate cancer cell motility and metastasis. Oncogene 2017, 36, 4597–4609. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.A.; Tan, S.-H.; Sun, C.; Shaheduzzaman, S.; Hu, Y.; Petrovics, G.; Chen, Y.; Sesterhenn, I.A.; Li, H.; Sreenath, T.; et al. ERG oncogene modulates prostaglandin signaling in prostate cancer cells. Cancer Biol. Ther. 2011, 11, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Klessig, D.F.; Tian, M.; Choi, H.W. Multiple Targets of Salicylic Acid and Its Derivatives in Plants and Animals. Front. Immunol. 2016, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shao, S.; Han, D.; Xu, Y.; Jiao, D.; Wu, J.; Yang, F.; Ge, Y.; Shi, S.; Li, Y.; et al. High mobility group box 1 promotes the epithelial-to-mesenchymal transition in prostate cancer PC3 cells via the RAGE/NF-κB signaling pathway. Int. J. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Tian, M.; Song, F.; Venereau, E.; Preti, A.; Park, S.-W.; Hamilton, K.; Swapna, G.V.T.; Manohar, M.; Moreau, M.; et al. Aspirin’s Active Metabolite Salicylic Acid Targets High Mobility Group Box 1 to Modulate Inflammatory Responses. Mol. Med. Camb. Mass 2015, 21, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Brusselaers, N. Maintenance use of aspirin or other non-steroidal anti-inflammatory drugs (NSAIDs) and prostate cancer risk. Prostate Cancer Prostatic Dis. 2018, 21, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Singh, S.; Shi, Q.; Pardee, A.B.; Iglehart, J.D. Crossroads of estrogen receptor and NF-kappaB signaling. Sci. STKE Signal Transduct. Knowl. Environ. 2005, 2005, pe27. [Google Scholar] [CrossRef]
- McKay, L.I.; Cidlowski, J.A. Cross-talk between nuclear factor-kappa B and the steroid hormone receptors: Mechanisms of mutual antagonism. Mol. Endocrinol. 1998, 12, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Nelius, T.; Filleur, S.; Yemelyanov, A.; Budunova, I.; Shroff, E.; Mirochnik, Y.; Aurora, A.; Veliceasa, D.; Xiao, W.; Wang, Z.; et al. Androgen receptor targets NFkappaB and TSP1 to suppress prostate tumor growth in vivo. Int. J. Cancer 2007, 121, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Palvimo, J.J.; Reinikainen, P.; Ikonen, T.; Kallio, P.J.; Moilanen, A.; Jänne, O.A. Mutual transcriptional interference between RelA and androgen receptor. J. Biol. Chem. 1996, 271, 24151–24156. [Google Scholar] [CrossRef] [PubMed]
- Maggio, M.; Basaria, S.; Ceda, G.P.; Ble, A.; Ling, S.M.; Bandinelli, S.; Valenti, G.; Ferrucci, L. The relationship between testosterone and molecular markers of inflammation in older men. J. Endocrinol. Investig. 2005, 28, 116–119. [Google Scholar]
- Zhang, B.; Kwon, O.-J.; Henry, G.; Malewska, A.; Wei, X.; Zhang, L.; Brinkley, W.; Zhang, Y.; Castro, P.D.; Titus, M.; et al. Non-Cell-Autonomous Regulation of Prostate Epithelial Homeostasis by Androgen Receptor. Mol. Cell 2016, 63, 976–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, L.; Saad, F.; Le Page, C.; Diallo, J.-S.; Péant, B.; Delvoye, N.; Mes-Masson, A.-M. NF-kappaB2 processing and p52 nuclear accumulation after androgenic stimulation of LNCaP prostate cancer cells. Cell. Signal. 2007, 19, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Campa, V.M.; Baltziskueta, E.; Bengoa-Vergniory, N.; Gorroño-Etxebarria, I.; Wesołowski, R.; Waxman, J.; Kypta, R.M. A screen for transcription factor targets of glycogen synthase kinase-3 highlights an inverse correlation of NFκB and androgen receptor signaling in prostate cancer. Oncotarget 2014, 5, 8173–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Nadiminty, N.; Liu, C.; Lou, W.; Schwartz, C.T.; Gao, A.C. Upregulation of glucose metabolism by NF-κB2/p52 mediates enzalutamide resistance in castration-resistant prostate cancer cells. Endocr. Relat. Cancer 2014, 21, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Nadiminty, N.; Tummala, R.; Liu, C.; Yang, J.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: Role of androgen receptor and its variants. Mol. Cancer Ther. 2013, 12, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Voogdt, C.; Tobias, A.; Spindler, K.-D.; Möller, P.; Cronauer, M.V.; Marienfeld, R.B. IκB kinases modulate the activity of the androgen receptor in prostate carcinoma cell lines. Neoplasia 2012, 14, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Mehraein-Ghomi, F.; Church, D.R.; Schreiber, C.L.; Weichmann, A.M.; Basu, H.S.; Wilding, G. Inhibitor of p52 NF-κB subunit and androgen receptor (AR) interaction reduces growth of human prostate cancer cells by abrogating nuclear translocation of p52 and phosphorylated AR(ser81). Genes Cancer 2015, 6, 428–444. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, N.K.; Ferdinand, A.S.; Mukhopadhyay, L.; Cinar, B.; Lutchman, M.; Richie, J.P.; Freeman, M.R.; Liu, B.C.-S. Unraveling androgen receptor interactomes by an array-based method: Discovery of proto-oncoprotein c-Rel as a negative regulator of androgen receptor. Exp. Cell Res. 2006, 312, 3782–3795. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, L.; McCall, P.; Hatziieremia, S.; Catlow, J.; Adams, C.; McArdle, P.; Seywright, M.; Tanahill, C.; Paul, A.; Underwood, M.; et al. Nuclear factor κB predicts poor outcome in patients with hormone-naive prostate cancer with high nuclear androgen receptor. Hum. Pathol. 2012, 43, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Ide, H.; Mizushima, T.; Jiang, G.; Netto, G.J.; Gotoh, M.; Miyamoto, H. Nuclear Factor-κB Promotes Urothelial Tumorigenesis and Cancer Progression via Cooperation with Androgen Receptor Signaling. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Malinen, M.; Niskanen, E.A.; Kaikkonen, M.U.; Palvimo, J.J. Crosstalk between androgen and pro-inflammatory signaling remodels androgen receptor and NF-κB cistrome to reprogram the prostate cancer cell transcriptome. Nucleic Acids Res. 2017, 45, 619–630. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Fu, Y.; Tian, D.; Yan, W. The contrasting roles of inflammasomes in cancer. Am. J. Cancer Res. 2018, 8, 566–583. [Google Scholar] [PubMed]
- Karan, D.; Tawfik, O.; Dubey, S. Expression analysis of inflammasome sensors and implication of NLRP12 inflammasome in prostate cancer. Sci. Rep. 2017, 7, 4378. [Google Scholar] [CrossRef] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like receptors and prostate cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Peek, E.M.; Song, W.; Zhang, H.; Huang, J.; Chin, A.I. Loss of MyD88 leads to more aggressive TRAMP prostate cancer and influences tumor infiltrating lymphocytes. Prostate 2015, 75, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Palchetti, S.; Starace, D.; De Cesaris, P.; Filippini, A.; Ziparo, E.; Riccioli, A. Transfected poly(I:C) activates different dsRNA receptors, leading to apoptosis or immunoadjuvant response in androgen-independent prostate cancer cells. J. Biol. Chem. 2015, 290, 5470–5483. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Zeng, C.; Xie, J.; Alghamdi, N.J.; Song, Y.; Zhang, H.; Zhou, A.; Jin, D. Effects of interferons and double-stranded RNA on human prostate cancer cell apoptosis. Oncotarget 2015, 6, 39184–39195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagna, M.; Takai, Y.; Kaibuchi, K.; Sano, K.; Kikkawa, U.; Nishizuka, Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J. Biol. Chem. 1982, 257, 7847–7851. [Google Scholar] [PubMed]
- De Weerd, A.; Kho, M.; Kraaijeveld, R.; Zuiderwijk, J.; Weimar, W.; Baan, C. The protein kinase C inhibitor sotrastaurin allows regulatory T cell function. Clin. Exp. Immunol. 2014, 175, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascher, A.; De Simone, P.; Pratschke, J.; Salamé, E.; Pirenne, J.; Isoneimi, H.; Bijarnia, M.; Krishnan, I.; Klupp, J. Protein kinase C inhibitor sotrastaurin in de novo liver transplant recipients: A randomized phase II trial. Am. J. Transplant. 2015, 15, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, D.; Williams, K.; LaFollette, T.D.; Cannon, M.; Blachly, J.S.; Zhong, Y.; Woyach, J.A.; Williams, E.; Awan, F.T.; Jones, J.; et al. PKC-β as a therapeutic target in CLL: PKC inhibitor AEB071 demonstrates preclinical activity in CLL. Blood 2014, 124, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Yokomizo, A.; Takeuchi, A.; Kashiwagi, E.; Dejima, T.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Eto, M. Protein kinase C regulates Twist1 expression via NF-κB in prostate cancer. Endocr. Relat. Cancer 2017, 24, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Totoń, E.; Ignatowicz, E.; Skrzeczkowska, K.; Rybczyńska, M. Protein kinase Cε as a cancer marker and target for anticancer therapy. Pharmacol. Rep. 2011, 63, 19–29. [Google Scholar] [CrossRef]
- Garg, R.; Blando, J.M.; Perez, C.J.; Abba, M.C.; Benavides, F.; Kazanietz, M.G. Protein Kinase C Epsilon Cooperates with PTEN Loss for Prostate Tumorigenesis through the CXCL13-CXCR5 Pathway. Cell Rep. 2017, 19, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Myatt, A.; Hill, S.J. Trypsin stimulates the phosphorylation of p42,44 mitogen-activated protein kinases via the proteinase-activated receptor-2 and protein kinase C epsilon in human cultured prostate stromal cells. Prostate 2005, 64, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.M.; Perez, C.J.; Lal, P.; Feldman, M.D.; Smyth, E.M.; Ricciotti, E.; Grosser, T.; Benavides, F.; Kazanietz, M.G. COX-2 mediates pro-tumorigenic effects of PKCε in prostate cancer. Oncogene 2018. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Gonzalez-Guerrico, A.; Kazanietz, M.G. PKC-mediated secretion of death factors in LNCaP prostate cancer cells is regulated by androgens. Mol. Carcinog. 2009, 48, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Pilling, A.B.; Hwang, O.; Boudreault, A.; Laurent, A.; Hwang, C. IAP Antagonists Enhance Apoptotic Response to Enzalutamide in Castration-Resistant Prostate Cancer Cells via Autocrine TNF-α Signaling. Prostate 2017, 77, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Win, H.Y.; Acevedo-Duncan, M. Atypical protein kinase C phosphorylates IKKalphabeta in transformed non-malignant and malignant prostate cell survival. Cancer Lett. 2008, 270, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, H.; Akimoto, K.; Nagashima, Y.; Kojima, Y.; Sasaki, T.; Ishiguro-Imagawa, Y.; Nakaigawa, N.; Ohno, S.; Kubota, Y.; Uemura, H. aPKClambda/iota promotes growth of prostate cancer cells in an autocrine manner through transcriptional activation of interleukin-6. Proc. Natl. Acad. Sci. USA 2009, 106, 16369–16374. [Google Scholar] [CrossRef] [PubMed]
- Campa, D.; Hüsing, A.; Stein, A.; Dostal, L.; Boeing, H.; Pischon, T.; Tjønneland, A.; Roswall, N.; Overvad, K.; Østergaard, J.N.; et al. Genetic variability of the mTOR pathway and prostate cancer risk in the European Prospective Investigation on Cancer (EPIC). PLoS ONE 2011, 6, e16914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cao, I.; Duran, A.; Collado, M.; Carrascosa, M.J.; Martín-Caballero, J.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J.; Serrano, M. Tumour-suppression activity of the proapoptotic regulator Par4. EMBO Rep. 2005, 6, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, H.-S.; Park, N.; Kwon, Y.-J.; Ye, D.-J.; Shin, S.; Chun, Y.-J. Annexin A5 suppresses cyclooxygenase-2 expression by downregulating the protein kinase C-ζ-nuclear factor-κB signaling pathway in prostate cancer cells. Oncotarget 2017, 8, 74263–74275. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Bee, A.; Brewer, D.; Dodson, A.; Beesley, C.; Ke, Y.; Ambroisine, L.; Fisher, G.; Møller, H.; Dickinson, T.; et al. PRKC-ζ Expression Promotes the Aggressive Phenotype of Human Prostate Cancer Cells and is a Novel Target for Therapeutic Intervention. Genes Cancer 2010, 1, 444–464. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Ireland, S.J.; Bee, A.; Beesley, C.; Forootan, S.S.; Dodson, A.; Dickinson, T.; Gerard, P.; Lian, L.-Y.; Risk, J.M.; et al. Splice variant PRKC-ζ(-PrC) is a novel biomarker of human prostate cancer. Br. J. Cancer 2012, 107, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Trani, M.; Sorrentino, A.; Busch, C.; Landström, M. Pro-apoptotic effect of aurothiomalate in prostate cancer cells. Cell Cycle 2009, 8, 306–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.-H.; Li, B.-X.; Han, Z.-P.; Zou, M.-X.; Wang, L.; Lv, Y.-B.; Zhou, J.-B.; Cao, M.-R.; Li, Y.-G.; Zhang, J.-Z. Snail-activated long non-coding RNA PCA3 up-regulates PRKD3 expression by miR-1261 sponging, thereby promotes invasion and migration of prostate cancer cells. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-S.; Melhuish, T.A.; Spencer, A.; Ni, L.; Hao, Y.; Jividen, K.; Harris, T.E.; Snow, C.; Frierson, H.F.; Wotton, D.; et al. The protein kinase C super-family member PKN is regulated by mTOR and influences differentiation during prostate cancer progression. Prostate 2017, 77, 1452–1467. [Google Scholar] [CrossRef] [PubMed]
- Jilg, C.A.; Ketscher, A.; Metzger, E.; Hummel, B.; Willmann, D.; Rüsseler, V.; Drendel, V.; Imhof, A.; Jung, M.; Franz, H.; et al. PRK1/PKN1 controls migration and metastasis of androgen-independent prostate cancer cells. Oncotarget 2014, 5, 12646–12664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickkholgh, B.; Sittadjody, S.; Rothberg, M.B.; Fang, X.; Li, K.; Chou, J.W.; Hawkins, G.A.; Balaji, K.C. Beta-catenin represses protein kinase D1 gene expression by non-canonical pathway through MYC/MAX transcription complex in prostate cancer. Oncotarget 2017, 8, 78811–78824. [Google Scholar] [CrossRef] [PubMed]
- Kaittanis, C.; Andreou, C.; Hieronymus, H.; Mao, N.; Foss, C.A.; Eiber, M.; Weirich, G.; Panchal, P.; Gopalan, A.; Zurita, J.; et al. Prostate-specific membrane antigen cleavage of vitamin B9 stimulates oncogenic signaling through metabotropic glutamate receptors. J. Exp. Med. 2017. [Google Scholar] [CrossRef]
- Rodriguez, M.; Siwko, S.; Liu, M. Prostate-Specific G-protein coupled receptor, an emerging biomarker regulating inflammation and prostate cancer invasion. Curr. Mol. Med. 2016, 16, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.S.; Lee, J.; Zhang, X.; Lindholm, P.F. Lysophosphatidic acid activates the RhoA and NF-κB through Akt/IκBα signaling and promotes prostate cancer invasion and progression by enhancing functional invadopodia formation. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 6775–6785. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.; Jung, Y.-J. Angiotensin II receptor blockers induce autophagy in prostate cancer cells. Oncol. Lett. 2017, 13, 3579–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, M.-T.; Wang, X.; Lee, D.T.; Tam, P.C.; Tsao, S.-W.; Wong, Y.-C. Id-1 expression induces androgen-independent prostate cancer cell growth through activation of epidermal growth factor receptor (EGF-R). Carcinogenesis 2004, 25, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Yowell, C.W.; Daaka, Y. G protein-coupled receptors provide survival signals in prostate cancer. Clin. Prostate Cancer 2002, 1, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Silvertown, J.D.; Ng, J.; Sato, T.; Summerlee, A.J.; Medin, J.A. H2 relaxin overexpression increases in vivo prostate xenograft tumor growth and angiogenesis. Int. J. Cancer 2006, 118, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Ishiguro, H.; Teranishi, J.-I.; Yoshida, S.-I.; Umemura, S.; Kubota, Y.; Uemura, H. Regulation of androgen receptor expression through angiotensin II type 1 receptor in prostate cancer cells. Prostate 2011, 71, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schuff-Werner, P.; Steiner, M. Thrombin/thrombin receptor (PAR-1)-mediated induction of IL-8 and VEGF expression in prostate cancer cells. Biochem. Biophys. Res. Commun. 2006, 343, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.M.; Brown, J.H. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol. Pharmacol. 2015, 88, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Tantivejkul, K.; Craig, M.; Neeley, C.K.; Pienta, K.J. PAR1-mediated RhoA activation facilitates CCL2-induced chemotaxis in PC-3 cells. J. Cell. Biochem. 2007, 101, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.L.; Mize, G.J.; Takayama, T.K. Protease-activated receptor mediated RhoA signaling and cytoskeletal reorganization in LNCaP cells. Biochemistry 2003, 42, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Vinall, R.L.; Mahaffey, C.M.; Davis, R.R.; Luo, Z.; Gandour-Edwards, R.; Ghosh, P.M.; Tepper, C.G.; de Vere White, R.W. Dual blockade of PKA and NF-κB inhibits H2 relaxin-mediated castrate-resistant growth of prostate cancer sublines and induces apoptosis. Horm. Cancer 2011, 2, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.-C.; Lin, M.-F. Protease-activated receptor 1: A role in prostate cancer metastasis. Clin. Prostate Cancer 2004, 3, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bastian, M.; Kohlschein, P.; Schuff-Werner, P.; Steiner, M. Expression of functional protease-activated receptor 1 in human prostate cancer cell lines. Urol. Res. 2003, 31, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, D.; Zhou, W.; Wang, M.; Xia, W.; Tang, Q. Prognostic value of matrix metalloprotease-1/protease-activated receptor-1 axis in patients with prostate cancer. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef] [PubMed]
- Black, P.C.; Mize, G.J.; Karlin, P.; Greenberg, D.L.; Hawley, S.J.; True, L.D.; Vessella, R.L.; Takayama, T.K. Overexpression of protease-activated receptors-1,-2, and-4 (PAR-1, -2, and -4) in prostate cancer. Prostate 2007, 67, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, V.; Kohli, M.; Dennis, R.A.; Siegel, E.R.; Chiles, W.W.; Mukunyadzi, P. Thrombin receptor expression is upregulated in prostate cancer. Prostate 2006, 66, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Al Saleh, H.A.; Haas-Neill, S.; Al-Hashimi, A.; Kapoor, A.; Shayegan, B.; Austin, R.C.; Al-Nedawi, K. Thrombotic characteristics of extracellular vesicles derived from prostate cancer cells. Prostate 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Mize, G.J.; Zhang, X.; Takayama, T.K. Kallikrein-related peptidase-4 initiates tumor-stroma interactions in prostate cancer through protease-activated receptor-1. Int. J. Cancer 2010, 126, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Nieman, M.T.; LaRusch, G.; Fang, C.; Zhou, Y.; Schmaier, A.H. Oral thrombostatin FM19 inhibits prostate cancer. Thromb. Haemost. 2010, 104, 1044–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekambaram, P.; Lee, J.-Y.L.; Hubel, N.E.; Hu, D.; Yerneni, S.; Campbell, P.G.; Pollock, N.; Klei, L.R.; Concel, V.J.; Delekta, P.C.; et al. The CARMA3-Bcl10-MALT1 Signalosome Drives NFκB Activation and Promotes Aggressiveness in Angiotensin II Receptor-Positive Breast Cancer. Cancer Res. 2018, 78, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P. A Role for the G12 Family of Heterotrimeric G Proteins in Prostate Cancer Invasion. J. Biol. Chem. 2006, 281, 26483–26490. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, X.; Wang, J.; Wang, Y.; Dong, H.; Li, J. Genetic variants in RhoA and ROCK1 genes are associated with the development, progression and prognosis of prostate cancer. Oncotarget 2017, 8, 19298–19309. [Google Scholar] [CrossRef] [PubMed]
- El-Haibi, C.P.; Sharma, P.; Singh, R.; Gupta, P.; Taub, D.D.; Singh, S.; Lillard, J.W. Differential G protein subunit expression by prostate cancer cells and their interaction with CXCR5. Mol. Cancer 2013, 12, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, P.M.; Bedolla, R.; Mikhailova, M.; Kreisberg, J.I. RhoA-dependent murine prostate cancer cell proliferation and apoptosis: Role of protein kinase Czeta. Cancer Res. 2002, 62, 2630–2636. [Google Scholar] [PubMed]
- Liu, Y.; Mo, J.Q.; Hu, Q.; Boivin, G.; Levin, L.; Lu, S.; Yang, D.; Dong, Z.; Lu, S. Targeted overexpression of vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer Res. 2008, 68, 6396–6406. [Google Scholar] [CrossRef] [PubMed]
- Péant, B.; Diallo, J.-S.; Lessard, L.; Delvoye, N.; Le Page, C.; Saad, F.; Mes-Masson, A.-M. Regulation of IkappaB kinase epsilon expression by the androgen receptor and the nuclear factor-kappaB transcription factor in prostate cancer. Mol. Cancer Res. 2007, 5, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Péant, B.; Gilbert, S.; Le Page, C.; Poisson, A.; L’Ecuyer, E.; Boudhraa, Z.; Bienz, M.N.; Delvoye, N.; Saad, F.; Mes-Masson, A.-M. IκB-Kinase-epsilon (IKKε) over-expression promotes the growth of prostate cancer through the C/EBP-β dependent activation of IL-6 gene expression. Oncotarget 2017, 8, 14487–14501. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Kreike, M.; Staal, J.; Beyaert, R. Importance of Validating Antibodies and Small Compound Inhibitors Using Genetic Knockout Studies-T Cell Receptor-Induced CYLD Phosphorylation by IKKε/TBK1 as a Case Study. Front. Cell Dev. Biol. 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Hutti, J.E.; Shen, R.R.; Abbott, D.W.; Zhou, A.Y.; Sprott, K.M.; Asara, J.M.; Hahn, W.C.; Cantley, L.C. Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKε promotes cell transformation. Mol. Cell 2009, 34, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Jeong, J.-H.; Park, S.-J.; Wei, R.; Peng, J.; Luo, Z.; Chen, Y.T.; Feng, Y.; Luo, J.-L. Selective TBK1/IKKi dual inhibitors with anticancer potency. Int. J. Cancer 2014, 134, 1972–1980. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, Q.; Liao, Q.; Wu, Q.; Sun, B.; Yang, Z.; Hu, X.; Tan, M.; Li, L. Interleukin-6/signal transducer and activator of transcription 3 promotes prostate cancer resistance to androgen deprivation therapy via regulating pituitary tumor transforming gene 1 expression. Cancer Sci. 2018, 109, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Jardin, S.E.; Kanchwala, M.S.; Jacob, J.; Merchant, S.; Meade, R.K.; Gahnim, N.M.; Nawas, A.F.; Xing, C.; Delk, N.A. Identification of an IL-1-induced gene expression pattern in AR+ PCa cells that mimics the molecular phenotype of AR- PCa cells. Prostate 2018, 78, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Shahriari, K.; Shen, F.; Worrede-Mahdi, A.; Liu, Q.; Gong, Y.; Garcia, F.U.; Fatatis, A. Cooperation among heterogeneous prostate cancer cells in the bone metastatic niche. Oncogene 2017, 36, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.A.; Patel, V.; Gwede, M.; Morgado, M.; Tomasevich, K.; Fong, E.L.; Farach-Carson, M.C.; Delk, N.A. IL-1β induces p62/SQSTM1 and represses androgen receptor expression in prostate cancer cells. J. Cell. Biochem. 2014, 115, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Russell, M.R.; Shahriari, K.; Jernigan, D.L.; Lioni, M.I.; Garcia, F.U.; Fatatis, A. Interleukin-1β promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res. 2013, 73, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Herroon, M.K.; Rajagurubandara, E.; Hardaway, A.L.; Powell, K.; Turchick, A.; Feldmann, D.; Podgorski, I. Bone marrow adipocytes promote tumor growth in bone via FABP4-dependent mechanisms. Oncotarget 2013, 4, 2108–2123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staverosky, J.A.; Zhu, X.-H.; Ha, S.; Logan, S.K. Anti-androgen resistance in prostate cancer cells chronically induced by interleukin-1β. Am. J. Clin. Exp. Urol. 2013, 1, 53–65. [Google Scholar] [PubMed]
- Chu, G.C.-Y.; Zhau, H.E.; Wang, R.; Rogatko, A.; Feng, X.; Zayzafoon, M.; Liu, Y.; Farach-Carson, M.C.; You, S.; Kim, J.; et al. RANK- and c-Met-mediated signal network promotes prostate cancer metastatic colonization. Endocr. Relat. Cancer 2014, 21, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Sircar, K.; Aprikian, A.; Potti, A.; Goltzman, D.; Rabbani, S.A. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer 2006, 107, 289–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, S.; Airoldi, I.; Sorrentino, C.; Zorzoli, A.; Esposito, S.; Di Carlo, E. Interleukin-30 expression in prostate cancer and its draining lymph nodes correlates with advanced grade and stage. Clin. Cancer Res. 2014, 20, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Omori, Y.; Lyn, D.; Singh, R.K.; Meinbach, D.M.; Sandman, Y.; Lokeshwar, V.B.; Lokeshwar, B.L. Interleukin-8 is a molecular determinant of androgen independence and progression in prostate cancer. Cancer Res. 2007, 67, 6854–6862. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Purcell, C.; Seaton, A.; Oladipo, O.; Maxwell, P.J.; O’Sullivan, J.M.; Wilson, R.H.; Johnston, P.G.; Waugh, D.J.J. Chemotherapy-induced CXC-chemokine/CXC-chemokine receptor signaling in metastatic prostate cancer cells confers resistance to oxaliplatin through potentiation of nuclear factor-kappaB transcription and evasion of apoptosis. J. Pharmacol. Exp. Ther. 2008, 327, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Roy, A.; Pahan, K. Selective neutralization of IL-12 p40 monomer induces death in prostate cancer cells via IL-12-IFN-γ. Proc. Natl. Acad. Sci. USA 2017, 114, 11482–11487. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Montgomery, R.B.; Schmidt, L.J.; Mostaghel, E.A.; Huang, H.; Nelson, P.S.; Tindall, D.J. Reduced tumor necrosis factor receptor-associated death domain expression is associated with prostate cancer progression. Cancer Res. 2009, 69, 9448–9456. [Google Scholar] [CrossRef] [PubMed]
- Galheigo, M.R.U.; Cruz, A.R.; Cabral, Á.S.; Faria, P.R.; Cordeiro, R.S.; Silva, M.J.B.; Tomiosso, T.C.; Gonçalves, B.F.; Pinto-Fochi, M.E.; Taboga, S.R.; et al. Role of the TNF-α receptor type 1 on prostate carcinogenesis in knockout mice. Prostate 2016, 76, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Berriguete, G.; Sánchez-Espiridión, B.; Cansino, J.R.; Olmedilla, G.; Martínez-Onsurbe, P.; Sánchez-Chapado, M.; Paniagua, R.; Fraile, B.; Royuela, M. Clinical significance of both tumor and stromal expression of components of the IL-1 and TNF-α signaling pathways in prostate cancer. Cytokine 2013, 64, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, E.; Staal, J.; Van Den Broeke, A.; Haegman, M.; Vereecke, L.; Schotte, P.; Beyaert, R. Prolonged exposure to IL-1beta and IFNgamma induces necrosis of L929 tumor cells via a p38MAPK/NF-kappaB/NO-dependent mechanism. Oncogene 2008, 27, 3780–3788. [Google Scholar] [CrossRef] [PubMed]
- Shelke, G.V.; Jagtap, J.C.; Kim, D.-K.; Shah, R.D.; Das, G.; Shivayogi, M.; Pujari, R.; Shastry, P. TNF-α and IFN-γ Together Up-Regulates Par-4 Expression and Induce Apoptosis in Human Neuroblastomas. Biomedicines 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, L.; Liu, H.; Duan, X.; Dickerson, E.; Shen, H.; Panchanathan, R.; Choubey, D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol. Cancer Res. 2013, 11, 1193–1202. [Google Scholar] [CrossRef] [PubMed]
- Lebel-Binay, S.; Thiounn, N.; De Pinieux, G.; Vieillefond, A.; Debré, B.; Bonnefoy, J.-Y.; Fridman, W.-H.; Pagès, F. IL-18 is produced by prostate cancer cells and secreted in response to interferons. Int. J. Cancer 2003, 106, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, B.W.-C.; Russell, P.J.; Lochner, M.; Förster, I.; Power, C.A. IL-18 inhibits growth of murine orthotopic prostate carcinomas via both adaptive and innate immune mechanisms. PLoS ONE 2011, 6, e24241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, S.; Goel, A.; Khattri, S.; Mandhani, A.; Sharma, P.; Misra, S.; Pant, K.K. Genetic variability at promoters of IL-18 (pro-) and IL-10 (anti-) inflammatory gene affects susceptibility and their circulating serum levels: An explorative study of prostate cancer patients in North Indian populations. Cytokine 2015, 74, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Jurecekova, J.; Babusikova, E.; Kmetova Sivonova, M.; Drobkova, H.; Petras, M.; Kliment, J.; Halasova, E. Association between interleukin-18 variants and prostate cancer in Slovak population. Neoplasma 2017, 64, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Liu, J.N.; Wei, M.T.; He, Y.Z.; Zhou, Y.; Song, X.B.; Ying, B.W.; Huang, J. Effect of IL-18 gene promoter polymorphisms on prostate cancer occurrence and prognosis in Han Chinese population. Genet. Mol. Res. GMR 2013, 12, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Saranchova, I.; Han, J.; Huang, H.; Fenninger, F.; Choi, K.B.; Munro, L.; Pfeifer, C.; Welch, I.; Wyatt, A.W.; Fazli, L.; et al. Discovery of a Metastatic Immune Escape Mechanism Initiated by the Loss of Expression of the Tumour Biomarker Interleukin-33. Sci. Rep. 2016, 6, 30555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, S.; Izumi, K.; Hiratsuka, K.; Maolake, A.; Natsagdorj, A.; Shigehara, K.; Iwamoto, H.; Kadomoto, S.; Makino, T.; Naito, R.; et al. C-C motif ligand 5 promotes migration of prostate cancer cells in the prostate cancer bone metastasis microenvironment. Cancer Sci. 2018, 109, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, J.; Xu, Y.; Koch, A.E.; Cai, Z.; Chen, X.; Galson, D.L.; Taichman, R.S.; Zhang, J. CXCL16 functions as a novel chemotactic factor for prostate cancer cells in vitro. Mol. Cancer Res. 2008, 6, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Schulze, J.; Weber, K.; Baranowsky, A.; Streichert, T.; Lange, T.; Spiro, A.S.; Albers, J.; Seitz, S.; Zustin, J.; Amling, M.; et al. p65-Dependent production of interleukin-1β by osteolytic prostate cancer cells causes an induction of chemokine expression in osteoblasts. Cancer Lett. 2012, 317, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Lou, W.; Nadiminty, N.; Lin, X.; Gao, A.C. Requirement for NF-(kappa)B in interleukin-4-induced androgen receptor activation in prostate cancer cells. Prostate 2005, 64, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Pinder, E.; Chun, J.Y.; Lou, W.; Sun, M.; Gao, A.C. Interleukin-4 stimulates androgen-independent growth in LNCaP human prostate cancer cells. Prostate 2008, 68, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Nappo, G.; Handle, F.; Santer, F.R.; McNeill, R.V.; Seed, R.I.; Collins, A.T.; Morrone, G.; Culig, Z.; Maitland, N.J.; Erb, H.H.H. The immunosuppressive cytokine interleukin-4 increases the clonogenic potential of prostate stem-like cells by activation of STAT6 signalling. Oncogenesis 2017, 6, e342. [Google Scholar] [CrossRef] [PubMed]
- Dluzniewski, P.J.; Wang, M.-H.; Zheng, S.L.; De Marzo, A.M.; Drake, C.G.; Fedor, H.L.; Partin, A.W.; Han, M.; Fallin, M.D.; Xu, J.; et al. Variation in IL10 and other genes involved in the immune response and in oxidation and prostate cancer recurrence. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2012, 21, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Takahashi, Y.; Narita, S.; Yang, Y.-C.; Li, X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget 2016, 8, 9375–9387. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staal, J.; Beyaert, R. Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells 2018, 7, 122. https://doi.org/10.3390/cells7090122
Staal J, Beyaert R. Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells. 2018; 7(9):122. https://doi.org/10.3390/cells7090122
Chicago/Turabian StyleStaal, Jens, and Rudi Beyaert. 2018. "Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications" Cells 7, no. 9: 122. https://doi.org/10.3390/cells7090122
APA StyleStaal, J., & Beyaert, R. (2018). Inflammation and NF-κB Signaling in Prostate Cancer: Mechanisms and Clinical Implications. Cells, 7(9), 122. https://doi.org/10.3390/cells7090122