The Ribosome Biogenesis—Cancer Connection



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
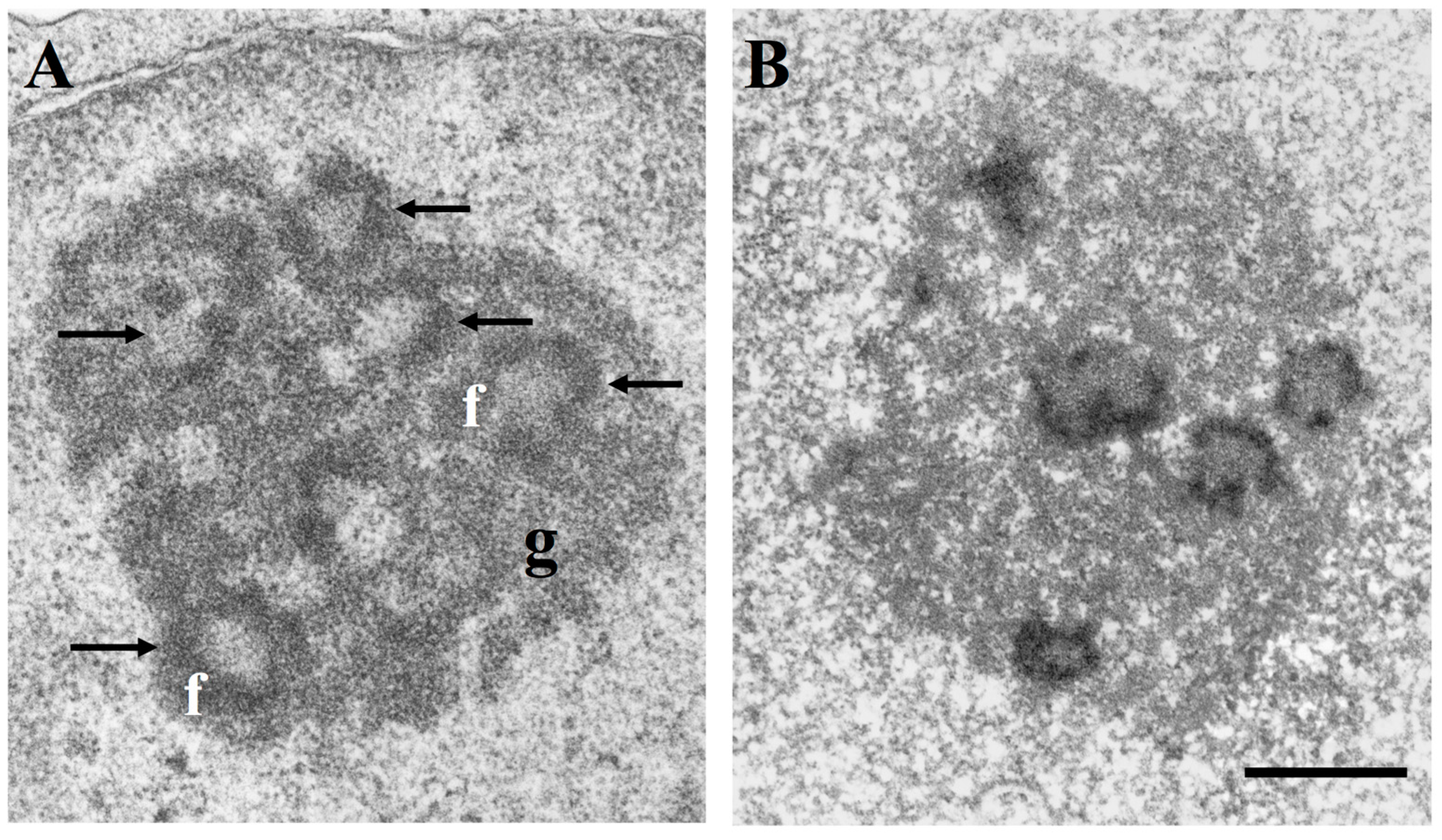
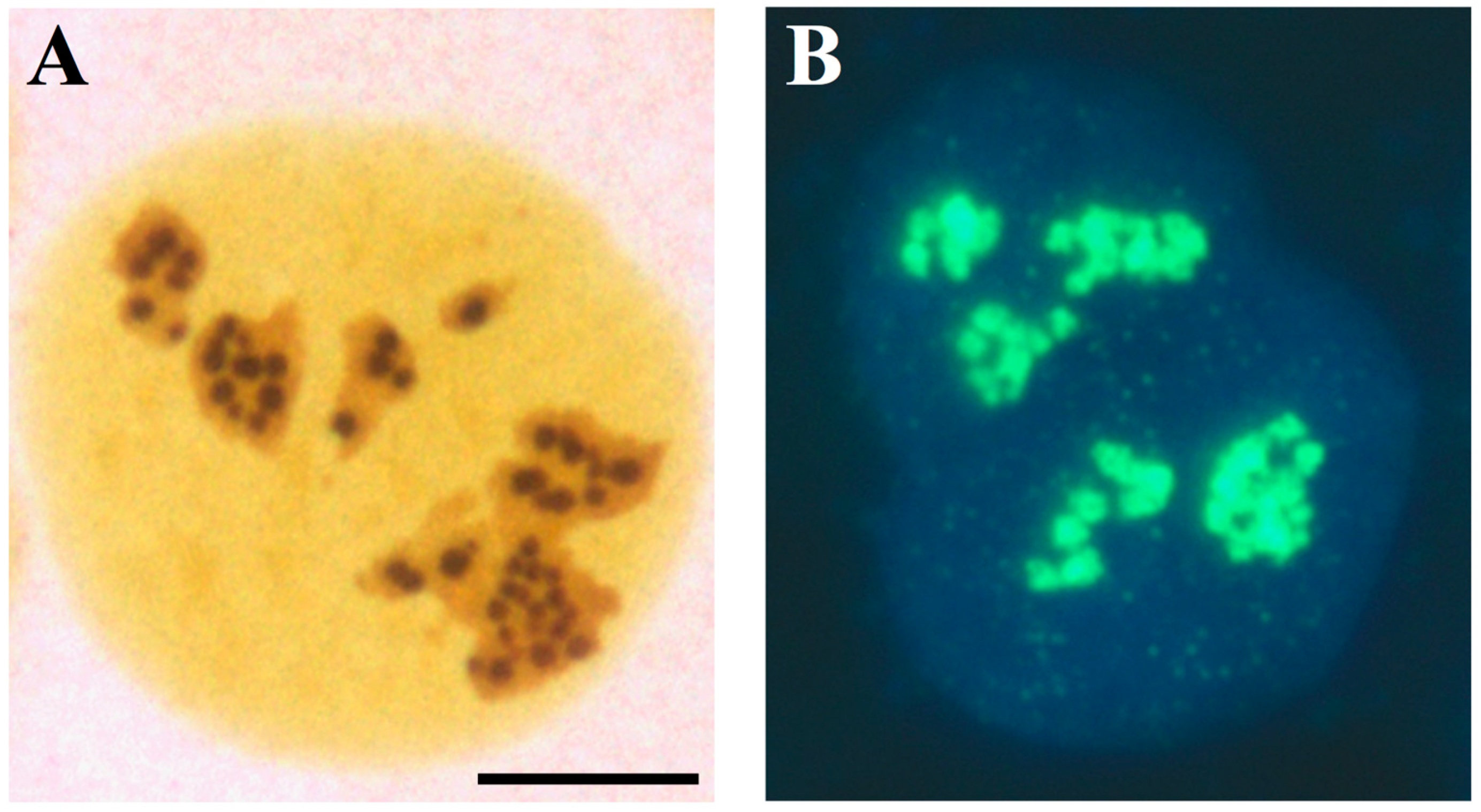
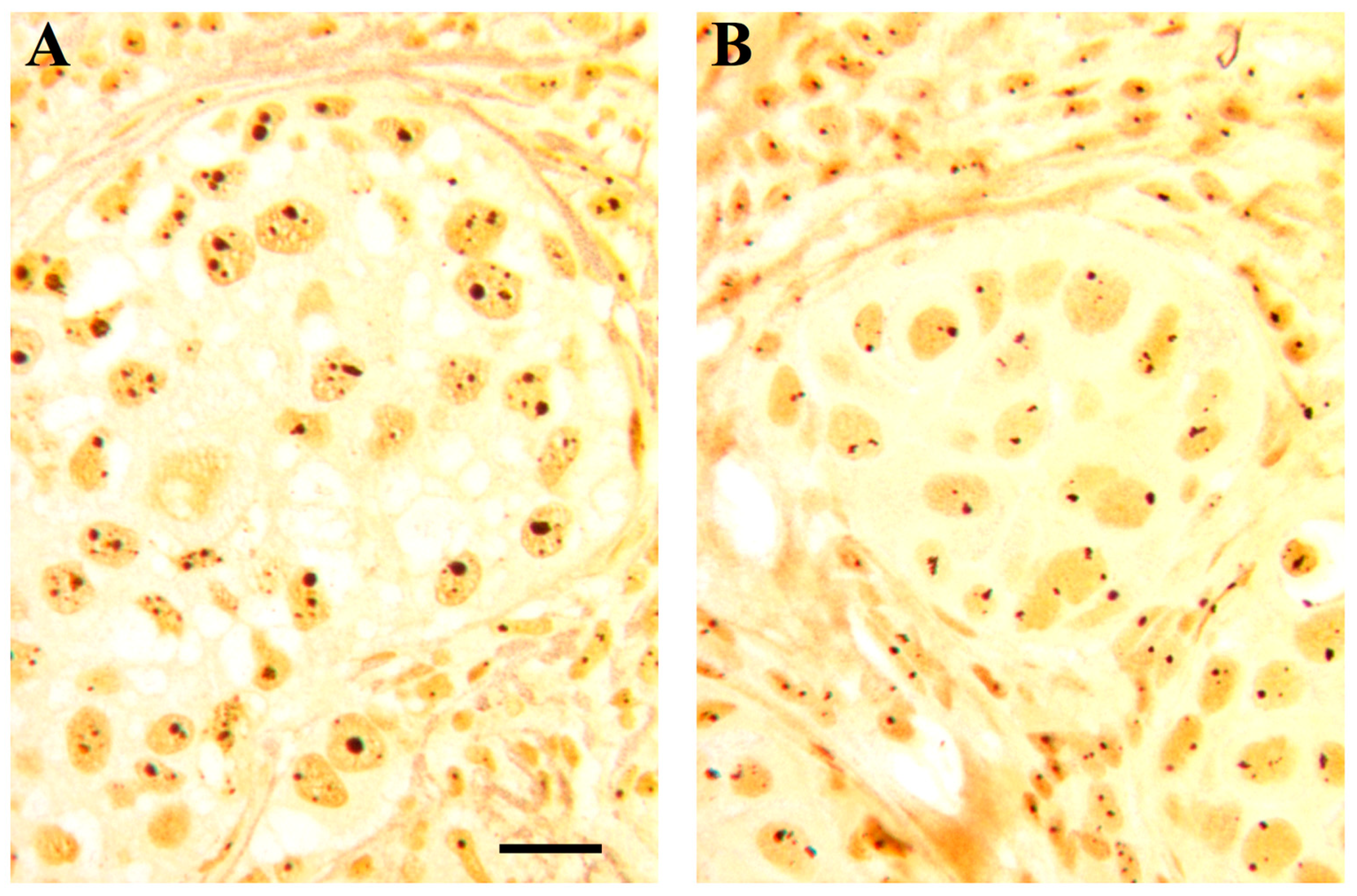
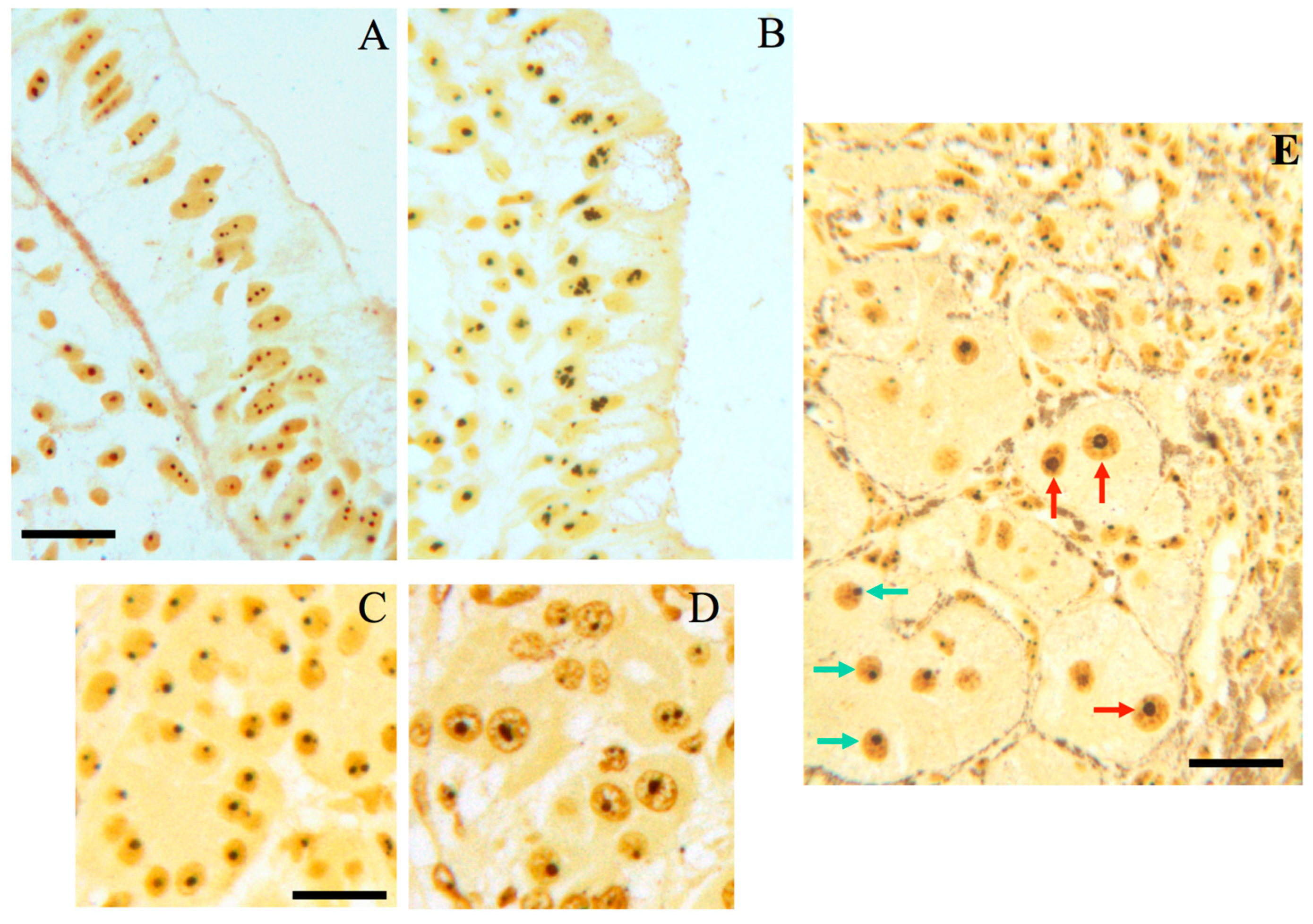
2. The Nucleolus in Tumor Pathology
3. Ribosome Biogenesis and Cancer Chemotherapy
3.1. Ribosome Biogenesis and Cell Cycle Regulation
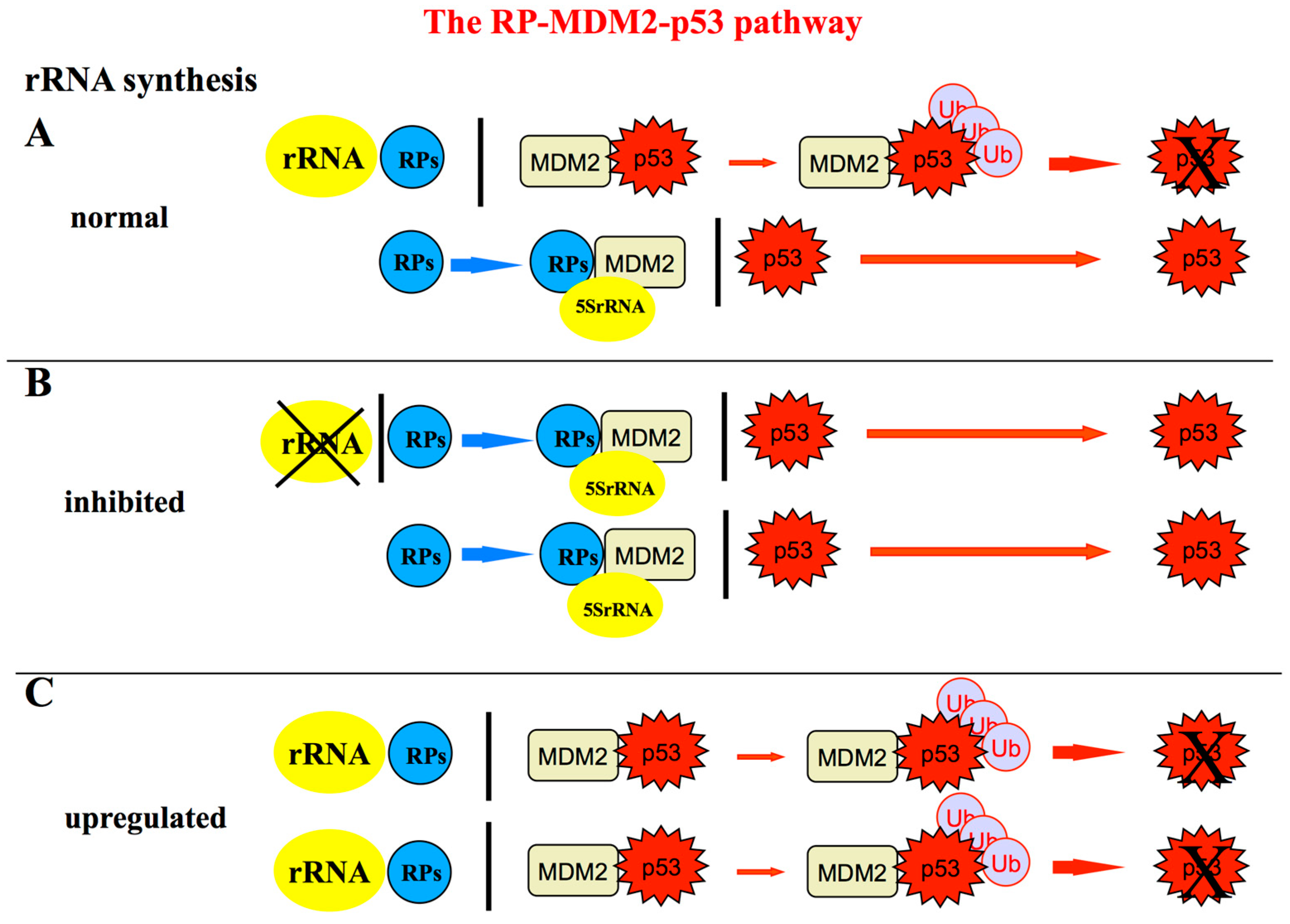
3.2. Ribosomal Stress and p53 Activation
3.3. Induction of Ribosomal Stress by Anticancer Agents
4. Ribosome Biogenesis and Neoplastic Transformation
4.1. Ribosome Biogenesis Rate and Cancer Development
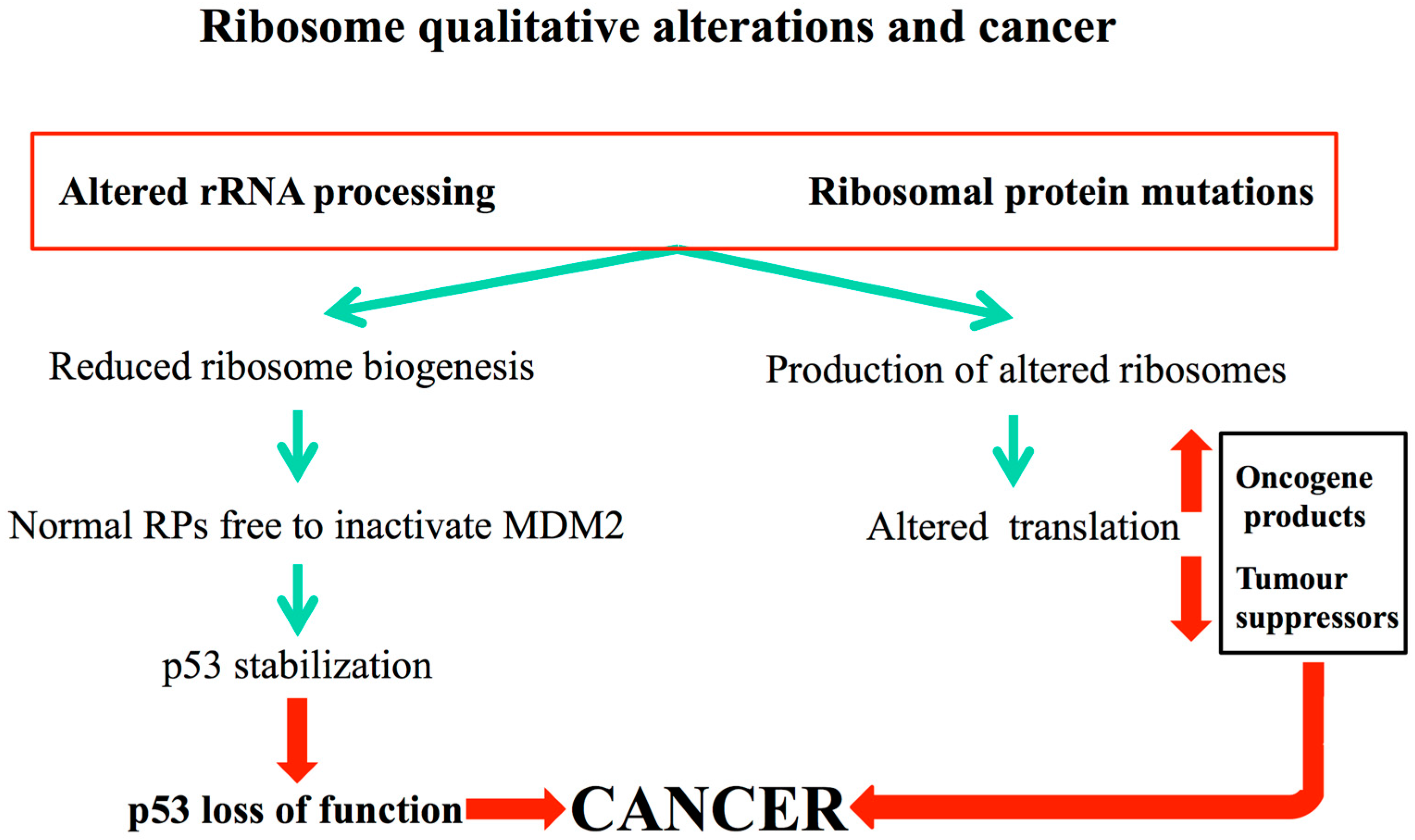
4.2. Qualitative Alterations in Ribosome Biogenesis and Cancer
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fatica, A.; Tollervey, D. Making ribosomes. Curr. Opin. Cell Biol. 2002, 14, 313–318. [Google Scholar] [CrossRef]
- Grummt, I. Life on a planet of its own: Regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003, 17, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Thiry, M.; Goessens, G. Ultrastructural cytochemistry of the mammalian cell nucleolus. J. Histochem. Cytochem. 1990, 38, 1237–1256. [Google Scholar] [CrossRef]
- Cisterna, B.; Biggiogera, M. Ribosome biogenesis: From structure to dynamics. Int. Rev. Cell Mol. Biol. 2010, 284, 67–111. [Google Scholar] [CrossRef]
- Derenzini, M.; Pasquinelli, G.; O’Donohue, M.-F.; Ploton, D.; Thiry, M. Structural and Functional Organization of Ribosomal Genes within the Mammalian Cell Nucleolus. J. Histochem. Cytochem. 2006, 54, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Verdun, D.; Roussel, P.; Thiry, M.; Sirri, V.; Lafontaine, D.L.J. The nucleolus: Structure/function relationship in RNA metabolism. Wiley Interdiscip. Rev. RNA 2010, 1, 415–431. [Google Scholar] [CrossRef]
- Raska, I.; Shaw, P.J.; Cmarko, D. Structure and function of the nucleolus in the spotlight. Curr. Opin. Cell Biol. 2006, 18, 325–334. [Google Scholar] [CrossRef]
- Lischwe, M.A.; Smetana, K.; Olson, M.O.; Busch, H. Proteins C23 and B23 are the major nucleolar silver staining proteins. Life Sci. 1979, 25, 701–708. [Google Scholar] [CrossRef]
- Hernandez-Verdun, D. Structural organization of the nucleolus in mammalian cells. Methods Achiev. Exp. Pathol. 1986, 12, 26–62. [Google Scholar] [PubMed]
- Derenzini, M.; Ploton, D. Interphase nucleolar organizer regions in cancer cells. Int. Rev. Exp. Pathol. 1991, 32, 149–192. [Google Scholar] [PubMed]
- Lindström, M.S.; Jurada, D.; Bursac, S.; Orsolic, I.; Bartek, J.; Volarevic, S. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 2018, 37, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pianese, G. Beitrag zur Histologie und Aetiologie der Carcinoma. Histologische und experimentelle Untersuchungen. Beitr. Pathol. Anat. Allg. Pathol. 1896, 142, 1–193. [Google Scholar]
- Ploton, D.; Menager, M.; Jeannesson, P.; Himber, G.; Pigeon, F.; Adnet, J.J. Improvement in the staining and in the visualization of the argyrophilic proteins of the nucleolar organizer region at the optical level. Histochem. J. 1986, 18, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Farabegoli, F.; Trerè, D. Relationship between interphase AgNOR distribution and nucleolar size in cancer cells. Histochem. J. 1992, 24, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Trerè, D.; Pession, A.; Montanaro, L.; Sirri, V.; Ochs, R.L. Nucleolar function and size in cancer cells. Am. J. Pathol. 1998, 152, 1291–1297. [Google Scholar] [PubMed]
- Trerè, D. AgNOR staining and quantification. Micron 2000, 31, 127–131. [Google Scholar] [CrossRef]
- Derenzini, M.; Trerè, D. Importance of interphase nucleolar organizer regions in tumor pathology. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1991, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Ploton, D. Interphase nucleolar organizer regions. In Molecular Biology in Histopathology; Crocker, J., Ed.; Wiley: New York, NY, USA, 1994; pp. 231–249. [Google Scholar]
- Derenzini, M. The AgNORs. Micron 2000, 31, 117–120. [Google Scholar] [CrossRef]
- Derenzini, M.; Montanaro, L.; Treré, D. What the nucleolus says to a tumour pathologist. Histopathology 2009, 54, 753–762. [Google Scholar] [CrossRef]
- Trerè, D.; Farabegoli, F.; Cancellieri, A.; Ceccarelli, C.; Eusebi, V.; Derenzini, M. AgNOR area in interphase nuclei of human tumours correlates with the proliferative activity evaluated by bromodeoxyuridine labelling and Ki-67 immunostaining. J. Pathol. 1991, 165, 53–59. [Google Scholar] [CrossRef]
- Derenzini, M.; Trerè, D.; Pession, A.; Govoni, M.; Sirri, V.; Chieco, P. Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J. Pathol. 2000, 191, 181–186. [Google Scholar] [CrossRef]
- Derenzini, M.; Sirri, V.; Pession, A.; Trerè, D.; Roussel, P.; Ochs, R.L.; Hernandez-Verdun, D. Quantitative Changes of the Two Major AgNOR Proteins, Nucleolin and Protein B23, Related to Stimulation of rDNA Transcription. Exp. Cell Res. 1995, 219, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, M.; Montanaro, L.; Chillà, A.; Tosti, E.; Vici, M.; Barbieri, S.; Govoni, M.; Mazzini, G.; Treré, D. Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J. Cell. Physiol. 2005, 202, 483–491. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 2005, 6, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roman, N.; Felton-Edkins, Z.A.; Kenneth, N.S.; Goodfellow, S.J.; Athineos, D.; Zhang, J.; Ramsbottom, B.A.; Innes, F.; Kantidakis, T.; Kerr, E.R.; et al. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem. Soc. Symp. 2006, 73, 141–154. [Google Scholar] [CrossRef]
- Van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Comai, L.; Johnson, D.L. Expression of PTEN in PTEN-deficient cells represses Pol I transcription by disrupting the SL1 complex. Mol. Cell. Biol. 2005, 25, 6899–6911. [Google Scholar] [CrossRef]
- Voit, R.; Schäfer, K.; Grummt, I. Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol. Cell. Biol. 1997, 17, 4230–4237. [Google Scholar] [CrossRef]
- Budde, A.; Grummt, I. p53 represses ribosomal gene transcription. Oncogene 1999, 18, 1119–1124. [Google Scholar] [CrossRef] [Green Version]
- Treré, D.; Ceccarelli, C.; Montanaro, L.; Tosti, E.; Derenzini, M. Nucleolar size and activity are related to pRb and p53 status in human breast cancer. J. Histochem. Cytochem. 2004, 52, 1601–1607. [Google Scholar] [CrossRef]
- Volarevic, S.; Stewart, M.J.; Ledermann, B.; Zilberman, F.; Terracciano, L.; Montini, E.; Grompe, M.; Kozma, S.C.; Thomas, G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science 2000, 288, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell. Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [PubMed]
- Barna, M.; Pusic, A.; Zollo, O.; Costa, M.; Kondrashov, N.; Rego, E.; Rao, P.H.; Ruggero, D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008, 456, 971–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherr, C.J. The Pezcoller lecture: Cancer cell cycles revisited. Cancer Res. 2000, 60, 3689–3695. [Google Scholar] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Flatt, P.M.; Tang, L.J.; Scatena, C.D.; Szak, S.T.; Pietenpol, J.A. p53 regulation of G(2) checkpoint is retinoblastoma protein dependent. Mol. Cell. Biol. 2000, 20, 4210–4223. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, G.; Brighenti, E.; Vici, M.; Mazzini, G.; Treré, D.; Montanaro, L.; Derenzini, M. Selective inhibition of rRNA transcription downregulates E2F-1: A new p53-independent mechanism linking cell growth to cell proliferation. J. Cell Sci. 2011, 124, 3017–3028. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Arnold, H.; Sun, X.-X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26, 3332–3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohrum, M.A.E.; Ludwig, R.L.; Kubbutat, M.H.G.; Hanlon, M.; Vousden, K.H. Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 2003, 3, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wolf, G.W.; Bhat, K.; Jin, A.; Allio, T.; Burkhart, W.A.; Xiong, Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 2003, 23, 8902–8912. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Lu, H. Inhibition of MDM2-mediated p53 Ubiquitination and Degradation by Ribosomal Protein L5. J. Biol. Chem. 2004, 279, 44475–44482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lu, H. Signaling to p53: Ribosomal proteins find their way. Cancer Cell 2009, 16, 369–377. [Google Scholar] [CrossRef]
- Bursac, S.; Brdovcak, M.C.; Donati, G.; Volarevic, S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Deisenroth, C.; Zhang, Y. Ribosome biogenesis surveillance: Probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 2010, 29, 4253–4260. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Fang, Z.; Liao, P.; Zhou, X.; Xiong, J.; Zeng, S.; Lu, H. Cancer-mutated ribosome protein L22 (RPL22/eL22) suppresses cancer cell survival by blocking p53-MDM2 circuit. Oncotarget 2017, 8, 90651–90661. [Google Scholar] [CrossRef]
- Bursać, S.; Brdovčak, M.C.; Pfannkuchen, M.; Orsolić, I.; Golomb, L.; Zhu, Y.; Katz, C.; Daftuar, L.; Grabušić, K.; Vukelić, I.; et al. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. USA 2012, 109, 20467–20472. [Google Scholar] [CrossRef] [Green Version]
- Donati, G.; Peddigari, S.; Mercer, C.A.; Thomas, G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013, 4, 87–98. [Google Scholar] [CrossRef]
- Sloan, K.E.; Bohnsack, M.T.; Watkins, N.J. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 2013, 5, 237–247. [Google Scholar] [CrossRef]
- Rubbi, C.P.; Milner, J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003, 22, 6068–6077. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [Green Version]
- Bruno, P.M.; Liu, Y.; Park, G.Y.; Murai, J.; Koch, C.E.; Eisen, T.J.; Pritchard, J.R.; Pommier, Y.; Lippard, S.J.; Hemann, M.T. A subset of platinum-containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat. Med. 2017, 23, 461–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [PubMed]
- Rickards, B.; Flint, S.J.; Cole, M.D.; LeRoy, G. Nucleolin Is Required for RNA Polymerase I Transcription In Vivo. Mol. Cell. Biol. 2007, 27, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Bywater, M.J.; Poortinga, G.; Sanij, E.; Hein, N.; Peck, A.; Cullinane, C.; Wall, M.; Cluse, L.; Drygin, D.; Anderes, K.; et al. Inhibition of RNA Polymerase I as a Therapeutic Strategy to Promote Cancer-Specific Activation of p53. Cancer Cell 2012, 22, 51–65. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Dos Santos, N.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef]
- Peltonen, K.; Colis, L.; Liu, H.; Jäämaa, S.; Moore, H.M.; Enbäck, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hällström, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef]
- Nikolov, E.N.; Dabeva, M.D.; Nikolov, T.K. Turnover of ribosomes in regenerating rat liver. Int. J. Biochem. 1983, 15, 1255–1260. [Google Scholar] [CrossRef]
- Stoykova, A.S.; Dudov, K.P.; Dabeva, M.D.; Hadjiolov, A.A. Different rates of synthesis and turnover of ribosomal RNA in rat brain and liver. J. Neurochem. 1983, 41, 942–949. [Google Scholar] [CrossRef]
- Scala, F.; Brighenti, E.; Govoni, M.; Imbrogno, E.; Fornari, F.; Treré, D.; Montanaro, L.; Derenzini, M. Direct relationship between the level of p53 stabilization induced by rRNA synthesis-inhibiting drugs and the cell ribosome biogenesis rate. Oncogene 2016, 35, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narla, A.; Ebert, B.L. Ribosomopathies: Human disorders of ribosome dysfunction. Blood 2010, 115, 3196–3205. [Google Scholar] [CrossRef] [PubMed]
- Trerè, D.; Borzio, M.; Morabito, A.; Borzio, F.; Roncalli, M.; Derenzini, M. Nucleolar hypertrophy correlates with hepatocellular carcinoma development in cirrhosis due to HBV infection. Hepatology 2003, 37, 72–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brighenti, E.; Calabrese, C.; Liguori, G.; Giannone, F.A.; Trerè, D.; Montanaro, L.; Derenzini, M. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: A new pathway connecting inflammation to cancer. Oncogene 2014, 33, 4396–4406. [Google Scholar] [CrossRef] [PubMed]
- Macchini, M. Relationship between Chronic Inflammation and Cancer: Interleukin-1β Overexpression Induces Pancreatic Ductal Adenocarcinoma in Oncogenic Kras Mice. Ph.D. Thesis, Alma Mater Studiorum Università di Bologna, Bologna, Italy, 2016. [Google Scholar]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.-Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Bertoni, S.; Brighenti, E.; Vici, M.; Treré, D.; Volarevic, S.; Montanaro, L.; Derenzini, M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene 2011, 30, 3274–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef]
- Yelick, P.C.; Trainor, P.A. Ribosomopathies: Global process, tissue specific defects. Rare Dis. 2015, 3, e1025185. [Google Scholar] [CrossRef] [Green Version]
- Farley, K.I.; Baserga, S.J. Probing the mechanisms underlying human diseases in making ribosomes. Biochem. Soc. Trans. 2016, 44, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Treré, D.; Derenzini, M. Changes in ribosome biogenesis may induce cancer by down-regulating the cell tumor suppressor potential. Biochim. Biophys. Acta 2012, 1825, 101–110. [Google Scholar] [CrossRef]
- Bellodi, C.; Krasnykh, O.; Haynes, N.; Theodoropoulou, M.; Peng, G.; Montanaro, L.; Ruggero, D. Loss of function of the tumor suppressor DKC1 perturbs p27 translation control and contributes to pituitary tumorigenesis. Cancer Res. 2010, 70, 6026–6035. [Google Scholar] [CrossRef]
- Bellodi, C.; Kopmar, N.; Ruggero, D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010, 29, 1865–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montanaro, L.; Calienni, M.; Bertoni, S.; Rocchi, L.; Sansone, P.; Storci, G.; Santini, D.; Ceccarelli, C.; Taffurelli, M.; Carnicelli, D.; et al. Novel Dyskerin-Mediated Mechanism of p53 Inactivation through Defective mRNA Translation. Cancer Res. 2010, 70, 4767–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocchi, L.; Pacilli, A.; Sethi, R.; Penzo, M.; Schneider, R.J.; Treré, D.; Brigotti, M.; Montanaro, L. Dyskerin depletion increases VEGF mRNA internal ribosome entry site-mediated translation. Nucleic Acids Res. 2013, 41, 8308–8318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penzo, M.; Rocchi, L.; Brugiere, S.; Carnicelli, D.; Onofrillo, C.; Coute, Y.; Brigotti, M.; Montanaro, L. Human ribosomes from cells with reduced dyskerin levels are intrinsically altered in translation. FASEB J. 2015, 29. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.W.; Green, R. Ribosomopathies: There’s strength in numbers. Science 2017, 358, eaan2755. [Google Scholar] [CrossRef]
- Gazda, H.T.; Sieff, C.A. Recent insights into the pathogenesis of Diamond?Blackfan anaemia. Br. J. Haematol. 2006, 135, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Dutt, S.; Narla, A.; Lin, K.; Mullally, A.; Abayasekara, N.; Megerdichian, C.; Wilson, F.H.; Currie, T.; Khanna-Gupta, A.; Berliner, N.; et al. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood 2011, 117, 2567–2576. [Google Scholar] [CrossRef]
- Elghetany, M.T.; Alter, B.P. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch. Pathol. Lab. Med. 2002, 126, 452–455. [Google Scholar] [CrossRef]
- Jones, N.C.; Lynn, M.L.; Gaudenz, K.; Sakai, D.; Aoto, K.; Rey, J.-P.; Glynn, E.F.; Ellington, L.; Du, C.; Dixon, J.; et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 2008, 14, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Fok, W.C.; de Oliveira Niero, E.L.; Dege, C.; Brenner, K.A.; Sturgeon, C.M.; Batista, L.F.Z. p53 Mediates Failure of Human Definitive Hematopoiesis in Dyskeratosis Congenita. Stem Cell Rep. 2017, 9, 409–418. [Google Scholar] [CrossRef]
- Marcel, V.; Nguyen Van Long, F.; Diaz, J.-J. 40 Years of Research Put p53 in Translation. Cancers 2018, 10, 152. [Google Scholar] [CrossRef]
- Ajore, R.; Raiser, D.; McConkey, M.; Jöud, M.; Boidol, B.; Mar, B.; Saksena, G.; Weinstock, D.M.; Armstrong, S.; Ellis, S.R.; et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with TP53 mutations. EMBO Mol. Med. 2017, 9, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancello, L.; Kampen, K.R.; Hofman, I.J.F.; Verbeeck, J.; De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 2017, 8, 14462–14478. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Lee, S.-Y.; Gutierrez, A.; Perrigoue, J.; Thapa, R.J.; Tu, Z.; Jeffers, J.R.; Rhodes, M.; Anderson, S.; Oravecz, T.; et al. Inactivation of ribosomal protein L22 promotes transformation by induction of the stemness factor, Lin28B. Blood 2012, 120, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novetsky, A.P.; Zighelboim, I.; Thompson, D.M.; Powell, M.A.; Mutch, D.G.; Goodfellow, P.J. Frequent mutations in the RPL22 gene and its clinical and functional implications. Gynecol. Oncol. 2013, 128, 470–474. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.M.; Tuominen, I.; van Dijk-Bos, K.; Sanjabi, B.; van der Sluis, T.; van der Zee, A.G.; Hollema, H.; Zazula, M.; Sijmons, R.H.; Aaltonen, L.A.; et al. High frequency of RPL22 mutations in microsatellite-unstable colorectal and endometrial tumors. Hum. Mutat. 2014, 35, 1442–1445. [Google Scholar] [CrossRef]
- Yang, M.; Sun, H.; Wang, H.; Zhang, S.; Yu, X.; Zhang, L. Down-regulation of ribosomal protein L22 in non-small cell lung cancer. Med. Oncol. 2013, 30, 646. [Google Scholar] [CrossRef]
- Nagarajan, N.; Bertrand, D.; Hillmer, A.M.; Zang, Z.J.; Yao, F.; Jacques, P.-É.; Teo, A.S.M.; Cutcutache, I.; Zhang, Z.; Lee, W.H.; et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012, 13, R115. [Google Scholar] [CrossRef]
- De Keersmaecker, K.; Atak, Z.K.; Li, N.; Vicente, C.; Patchett, S.; Girardi, T.; Gianfelici, V.; Geerdens, E.; Clappier, E.; Porcu, M.; et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat. Genet. 2012, 45, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Brighenti, E.; Giannone, F.A.; Fornari, F.; Onofrillo, C.; Govoni, M.; Montanaro, L.; Treré, D.; Derenzini, M. Therapeutic dosages of aspirin counteract the IL-6 induced pro-tumorigenic effects by slowing down the ribosome biogenesis rate. Oncotarget 2016, 7, 63226–63241. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Penzo, M.; Montanaro, L.; Treré, D.; Derenzini, M. The Ribosome Biogenesis—Cancer Connection. Cells 2019, 8, 55. https://doi.org/10.3390/cells8010055
Penzo M, Montanaro L, Treré D, Derenzini M. The Ribosome Biogenesis—Cancer Connection. Cells. 2019; 8(1):55. https://doi.org/10.3390/cells8010055
Chicago/Turabian StylePenzo, Marianna, Lorenzo Montanaro, Davide Treré, and Massimo Derenzini. 2019. "The Ribosome Biogenesis—Cancer Connection" Cells 8, no. 1: 55. https://doi.org/10.3390/cells8010055
APA StylePenzo, M., Montanaro, L., Treré, D., & Derenzini, M. (2019). The Ribosome Biogenesis—Cancer Connection. Cells, 8(1), 55. https://doi.org/10.3390/cells8010055