Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines

 , ,
, ,
Abstract
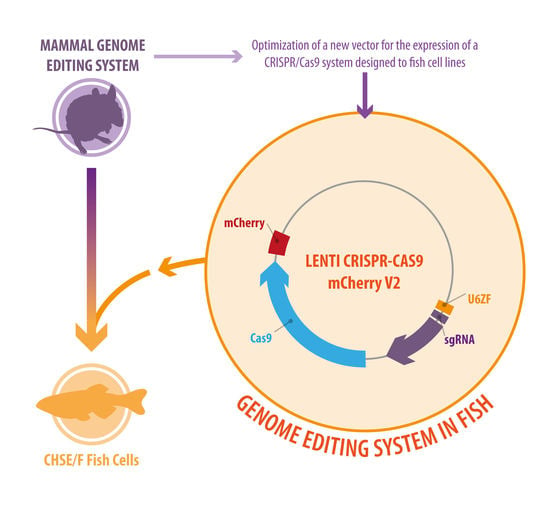
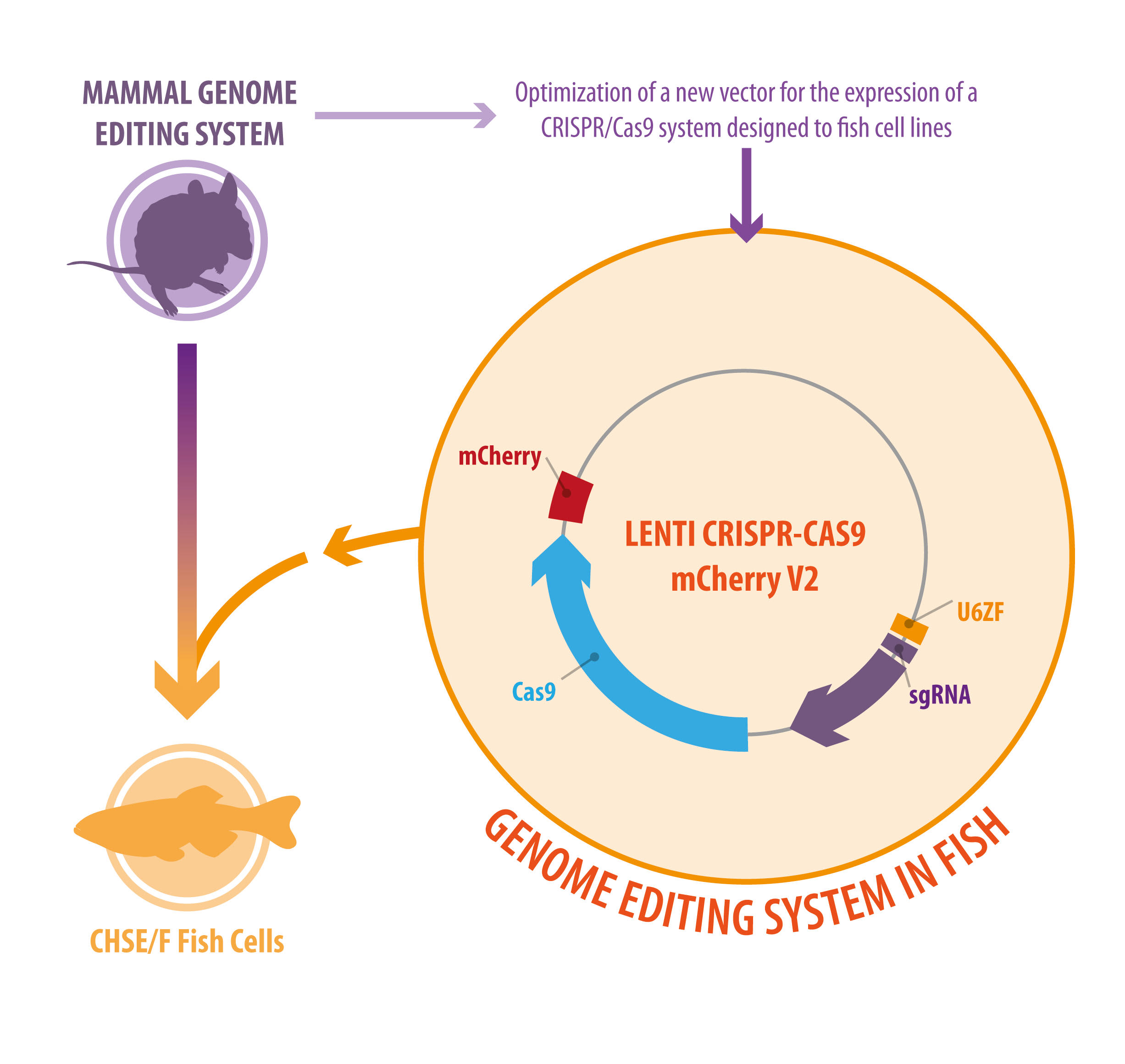
:
1. Introduction
2. Materials and Methods
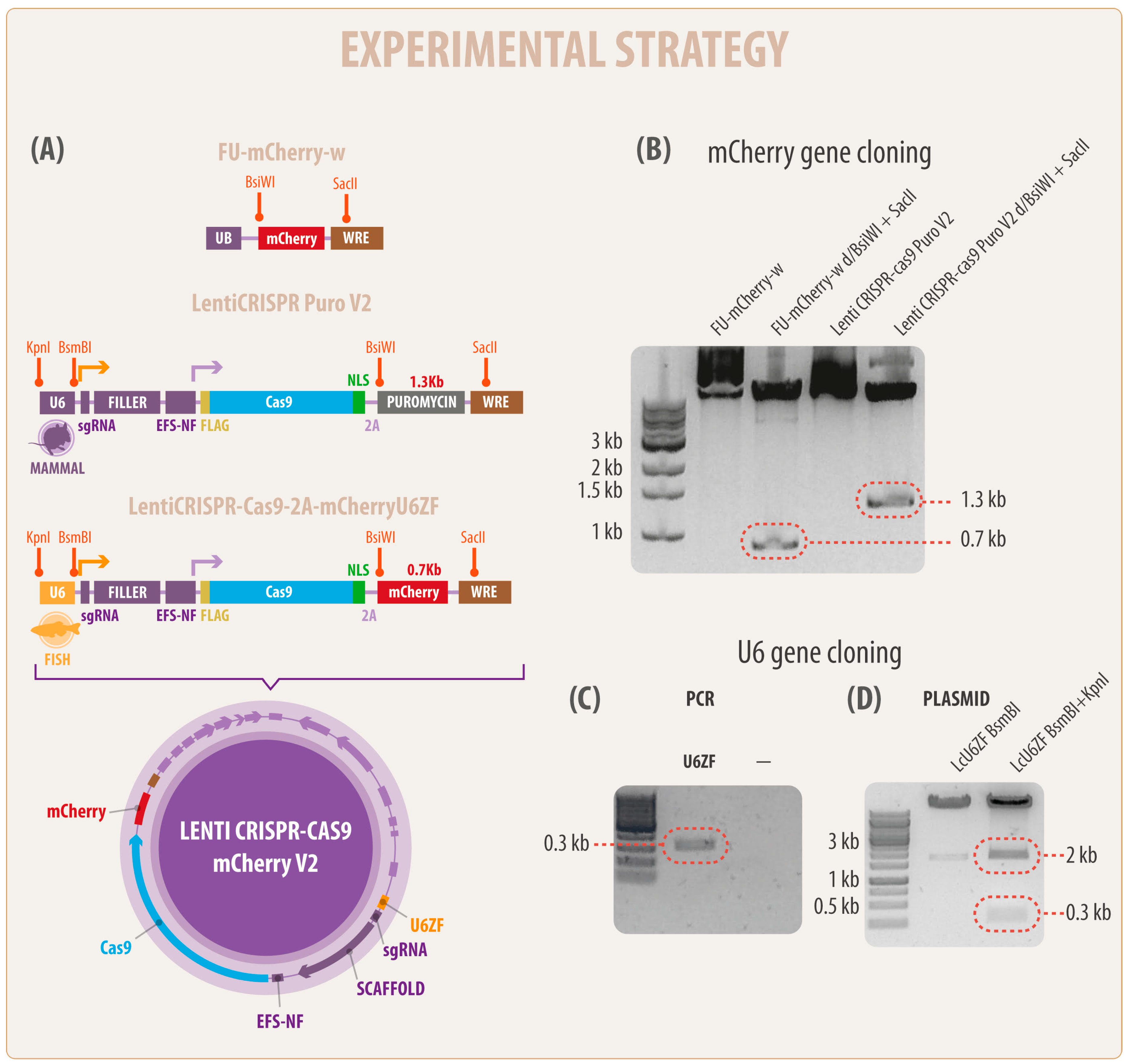
2.1. Plasmid Vector Construction
2.2. Cloning sgRNA Oligonucleotide in the Novel LcU6ZF Vector
2.3. Cell Culture and Rates of Transfection
2.4. RNA Isolation and Reverse Transcription (cDNA) of CHSE/F Cells
2.5. PCR of CHSE/F Cells Transfected by LcU6ZFsgGFP
2.6. Fluorescent Monolayer of Transfected CHSE/F Cells
2.7. Western Blotting
2.8. Genome Editing on Human Cell Lines Transfected with a Novel LcU6ZF Plasmid
2.9. Cell Sorting of CHSE/F Cell Lines Transfected with LcU6ZF β-Actin Plasmid
2.10. High Resolution Melting Analysis of Genome-Edited CHSE/F Cells
3. Results
3.1. Generation of a Novel Bicistronic (Construct) CRISPR/Cas9 System for Modification of Fish Cell Lines
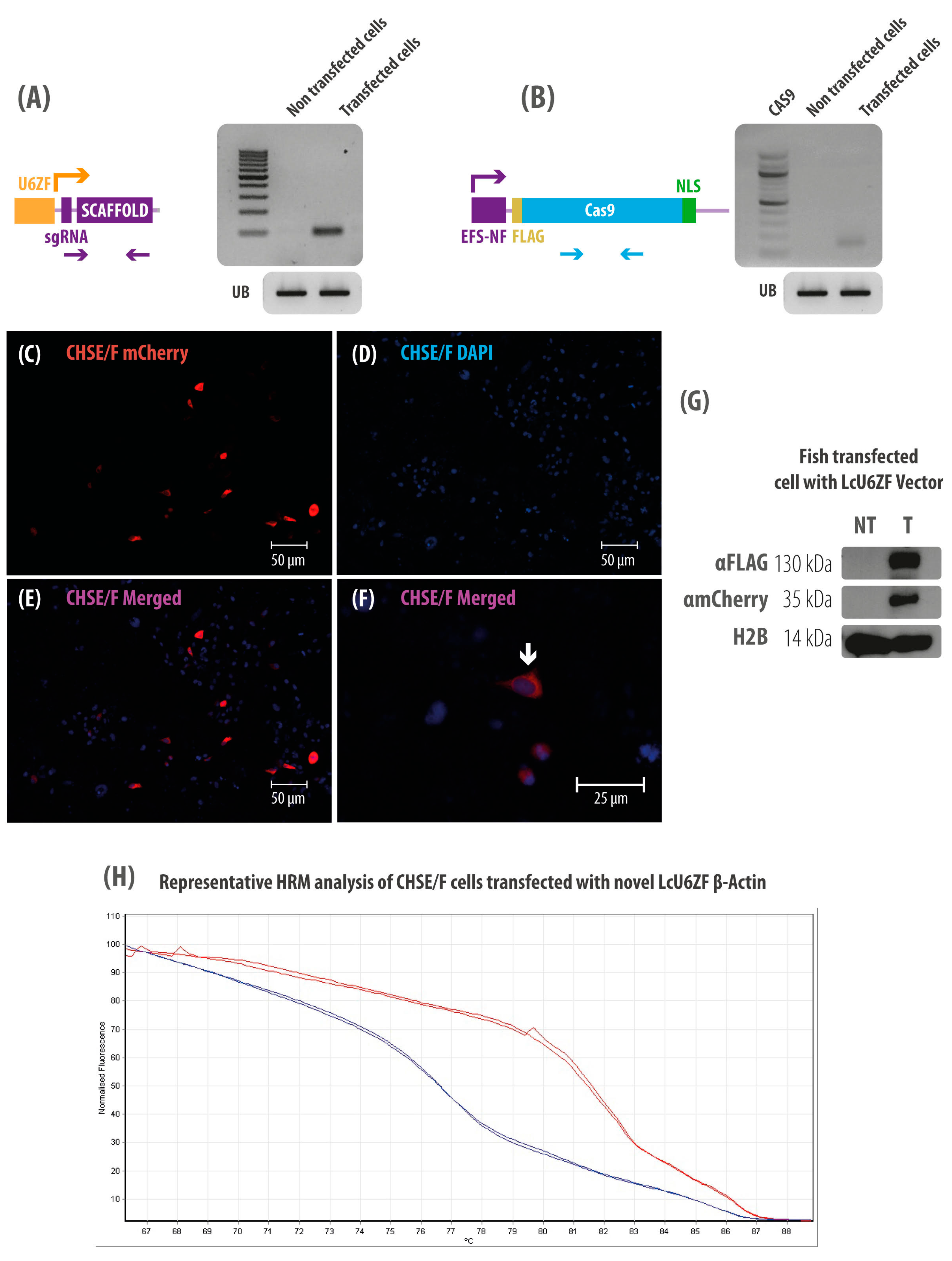
3.2. CHSE/F Transfected Cells Expressing both sgRNA and Cas9
3.3. CHSE/F Transfected Cells Express mCherry and Cas9 Protein
3.4. Human Cell Knocked-Out CDNF Protein Using the Novel Vector LcU6ZF
3.5. Detecting Single-Base Mutations in the Genome of CHSE/F Cells Using LcU6ZF β-Actin Vector by HRM Curve Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.R.J.; Joung, J.K. Efficient in vivo genome editing using RNA-guided nucleases. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Dong, F.; Xie, K.; Chen, Y.; Yang, Y.; Mao, Y. Polycistronic tRNA and CRISPR guide-RNA enables highly efficient multiplexed genome engineering in human cells. Biochem. Biophys. Res. Commun. 2017, 482, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for genome editing, regulation and targeting. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Liu, F.; Gu, P.; Jin, H.; Chang, Y.; Wang, Q.; Liang, Q.; Qi, Q. A CRISPR-Cas9 assisted non-homologous end-joining strategy for one-step engineering of bacterial genome. Sci. Rep. 2016, 6, 37895. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edvardsen, R.B.; Leininger, S.; Kleppe, L.; Skaftnesmo, K.O.; Wargelius, A. Targeted mutagenesis in Atlantic Salmon (Salmo salar L.) using the CRISPR/Cas9 system induces complete knockout individuals in the F0 generation. PLoS ONE 2014, 9, e108622. [Google Scholar] [CrossRef]
- Li, M.; Yang, H.; Zhao, J.; Fang, L.; Shi, H.; Li, M.; Sun, Y.; Zhang, X.; Jiang, D.; Zhou, L.; et al. Efficient and heritable gene targeting in Tilapia by CRISPR/Cas9. Genetics 2014, 197, 591–599. [Google Scholar] [CrossRef]
- Feng, R.; Fang, L.; Cheng, Y.; He, X.; Jiang, W.; Dong, R.; Shi, H.; Jiang, D.; Sun, L.; Wang, D. Retinoic acid homeostasis through aldh1a2 and cyp26a1 mediates meiotic entry in Nile tilapia (Oreochromis niloticus). Sci. Rep. 2015, 5, 10131. [Google Scholar] [CrossRef]
- Wargelius, A.; Leininger, S.; Skaftnesmo, K.O.; Kleppe, L.; Andersson, E.; Taranger, G.L.; Schulz, R.W.; Edvardsen, R.B. Dnd knockout ablates germ cells and demonstrates germ cell independent sex differentiation in Atlantic salmon. Sci. Rep. 2016, 6, 21284. [Google Scholar] [CrossRef]
- Ansai, S.; Kinoshita, M. Targeted mutagenesis using CRISPR/Cas system in medaka. Biol. Open 2014, 3, 362–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehler, C.E.; Boudinot, P.; Martin, S.A.M.; Collet, B. Development of an efficient genome editing method by CRISPR/Cas9 in a fish cell line. Mar. Biotechnol. 2016, 18, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Yuan, Y.; Zhu, F.; Hong, Y.; Ge, R. Efficient genome editing using CRISPR/Cas9 ribonucleoprotein approach in cultured Medaka fish cells. Biol. Open 2018, 7, bio035170. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lois, C.; Hong, E.J.; Pease, S.; Brown, E.J.; Baltimore, D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 2002, 295, 868–872. [Google Scholar] [CrossRef]
- Shinya, M.; Kobayashi, K.; Masuda, A.; Tokumoto, M.; Ozaki, Y.; Saito, K.; Kawasaki, T.; Sado, Y.; Sakai, N. Properties of gene knockdown system by vector-based siRNA in zebrafish. Dev. Growth Differ. 2013, 55, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Peña, A.A.; Bols, N.C.; Marshall, S.H. An evaluation of potential reference genes for stability of expression in two salmonid cell lines after infection with either Piscirickettsia salmonis or IPNV. BMC Res. Notes 2010, 3, 101. [Google Scholar] [CrossRef]
- Wittwer, C.T. High-resolution DNA melting analysis: advancements and limitations. Hum. Mutat. 2009, 30, 857–859. [Google Scholar] [CrossRef] [Green Version]
- Bearzotti, M.; Perrot, E.; Michard-Vanhee, C.; Jolivet, G.; Attal, J.; Theron, M.C.; Puissant, C.; Dreano, M.; Kopchick, J.J.; Powell, R.; et al. Gene expression following transfection of fish cells. J. Biotechnol. 1992, 26, 315–325. [Google Scholar] [CrossRef]
- Pitkänen, T.I.; Krasnov, A.; Teerijoki, H.; Mölsä, H. Transfer of growth hormone (GH) transgenes into Arctic charr (Salvelinus alpinus L.): I. Growth response to various GH constructs. Genet. Anal. Biomol. Eng. 1999, 15, 91–98. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Yoshizaki, G.; Takeuchi, T. Green fluorescent protein as a cell-labeling tool and a reporter of gene expression in transgenic rainbow trout. Mar. Biotechnol. 1999, 1, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Villalobos, P.; Rojas, M.V.; Conejeros, P.; Marshall, S.H. Lipopolyamine-mediated transfection of reporter plasmids into a fish cell line. Electron. J. Biotechnol. 1999, 2, 2. [Google Scholar] [CrossRef]
- Deng, W.; Yang, D.; Zhao, B.; Ouyang, Z.; Song, J.; Fan, N.; Liu, Z.; Zhao, Y.; Wu, Q.; Nashun, B.; et al. Use of the 2A peptide for generation of multi-transgenic pigs through a single round of nuclear transfer. PLoS ONE 2011, 6, e19986. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, S.R.; Li, L.H.; Park, J.H.; Lee, K.Y.; Kim, M.K.; Shin, B.A.; Choi, S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE 2011, 6, e18556. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184. [Google Scholar] [CrossRef] [PubMed]
- Pshenichkin, S.; Surin, A.; Surina, E.; Klauzińska, M.; Grajkowska, E.; Luchenko, V.; Dolińska, M.; Wroblewska, B.; Wroblewski, J.T. Heat shock enhances CMV-IE promoter-driven metabotropic glutamate receptor expression and toxicity in transfected cells. Neuropharmacology 2011, 60, 1292–1300. [Google Scholar] [CrossRef] [Green Version]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, R.; Zhao, P.; Xia, Q. 2A self-cleaving peptide-based multi-gene expression system in the silkworm Bombyx mori. Sci. Rep. 2015, 5, 16273. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.H.; Liang, Y.S.; Jung, H.; Ahn, M.J.; Suh, S.C.; Kweon, S.J.; Kim, D.H.; Kim, Y.M.; Kim, J.K. Application of two bicistronic systems involving 2A and IRES sequences to the biosynthesis of carotenoids in rice endosperm. Plant Biotechnol. J. 2010, 8, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Detleff, P.; Hernandez, E.; Martinez, V. A comprehensive transcriptome of early development in yellowtail kingfish (Seriola lalandi). Mol. Ecol. Resour. 2016, 16, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Samarut, E.; Lissouba, A.; Drapeau, P. A simplified method for identifying early CRISPR-induced indels in zebrafish embryos using High Resolution Melting analysis. BMC Genomics 2016, 17, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Name | Sequence 5′–3′ |
|---|---|
| U6ZF_F [16] | GTGTGGTACCACCTCAACAAAAGCTCCTCGATGT |
| U6F_R [16] | CAACCGTCTCCGGTGTGGGAGTCTGGAGGACGGCTATATA |
| GFPA | CACCGGGTGAACCGCATCGAGCTGA |
| GFPB | AAACTCAGCTCGATGCGGTTCACCC |
| Ubq_F [17] | GGAAAACCATCACCCTTGAG |
| Ubq_R [17] | ATAATGCCTCCACGAAGACG |
| FwdGFPPCR | GGTGAACCGCATCGAGCTGA |
| RvsgRNAscaffold | ACCGACTCGGTGCCACTTTT |
| sgRNA1CDNF-A | CACCGACTTGGCGTCGGTGGACCTG |
| sgRNA1CDNF-B | AAACCAGGTCCACCGACGCCAAGTCC |
| sgRNA2CDNF-A | CACCTTGTATCTCGAACCCTGTGC |
| sgRNA2CDNF-B | AAACGCACAGGGTTCGAGATACAAC |
| sgRNAβactin-A | CACCGCGCCGGAGATGACGCGCCTC |
| sgRNAβactin-B | AAACGAGGCGCGTCATCTCCGGCGC |
| βActin HRM-Fwd | GGATCCGGTATGTGCAAAGCC |
| βActin HRM-Rv | CGTCCCAAAGCCCATCATGAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escobar-Aguirre, S.; Arancibia, D.; Escorza, A.; Bravo, C.; Andrés, M.E.; Zamorano, P.; Martínez, V. Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines. Cells 2019, 8, 75. https://doi.org/10.3390/cells8010075
Escobar-Aguirre S, Arancibia D, Escorza A, Bravo C, Andrés ME, Zamorano P, Martínez V. Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines. Cells. 2019; 8(1):75. https://doi.org/10.3390/cells8010075
Chicago/Turabian StyleEscobar-Aguirre, Sebastian, Duxan Arancibia, Amanda Escorza, Cristián Bravo, María Estela Andrés, Pedro Zamorano, and Víctor Martínez. 2019. "Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines" Cells 8, no. 1: 75. https://doi.org/10.3390/cells8010075
APA StyleEscobar-Aguirre, S., Arancibia, D., Escorza, A., Bravo, C., Andrés, M. E., Zamorano, P., & Martínez, V. (2019). Development of a Bicistronic Vector for the Expression of a CRISPR/Cas9-mCherry System in Fish Cell Lines. Cells, 8(1), 75. https://doi.org/10.3390/cells8010075