Phosphoinositide 3-Kinase P110δ-Signaling Is Critical for Microbiota-Activated IL-10 Production by B Cells that Regulate Intestinal Inflammation

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Cecal Bacterial Lysate (CBL)
2.3. Cell Isolation from Spleen, MLN and cLP
2.4. Cell Purification of APCs, BMDMs, B Cells and Naïve T Cells
2.5. Cell Culture with Bacterial Products and PI3K-Related Inhibitors
2.6. B Cell Co-Culture Assays with CD4+ T Cells and Macrophages
2.7. T cell Proliferation Assay
2.8. Adoptive Transfer of Naïve CD4+ T Cells and CD19+ B Cells
2.9. Preparation for Quantification of Fecal Lipocalin-2
2.10. ELISA
2.11. Quantitative PCR
2.12. Flow Cytometry
2.13. Histological Colitis Score
2.14. Statistical Analysis
3. Results
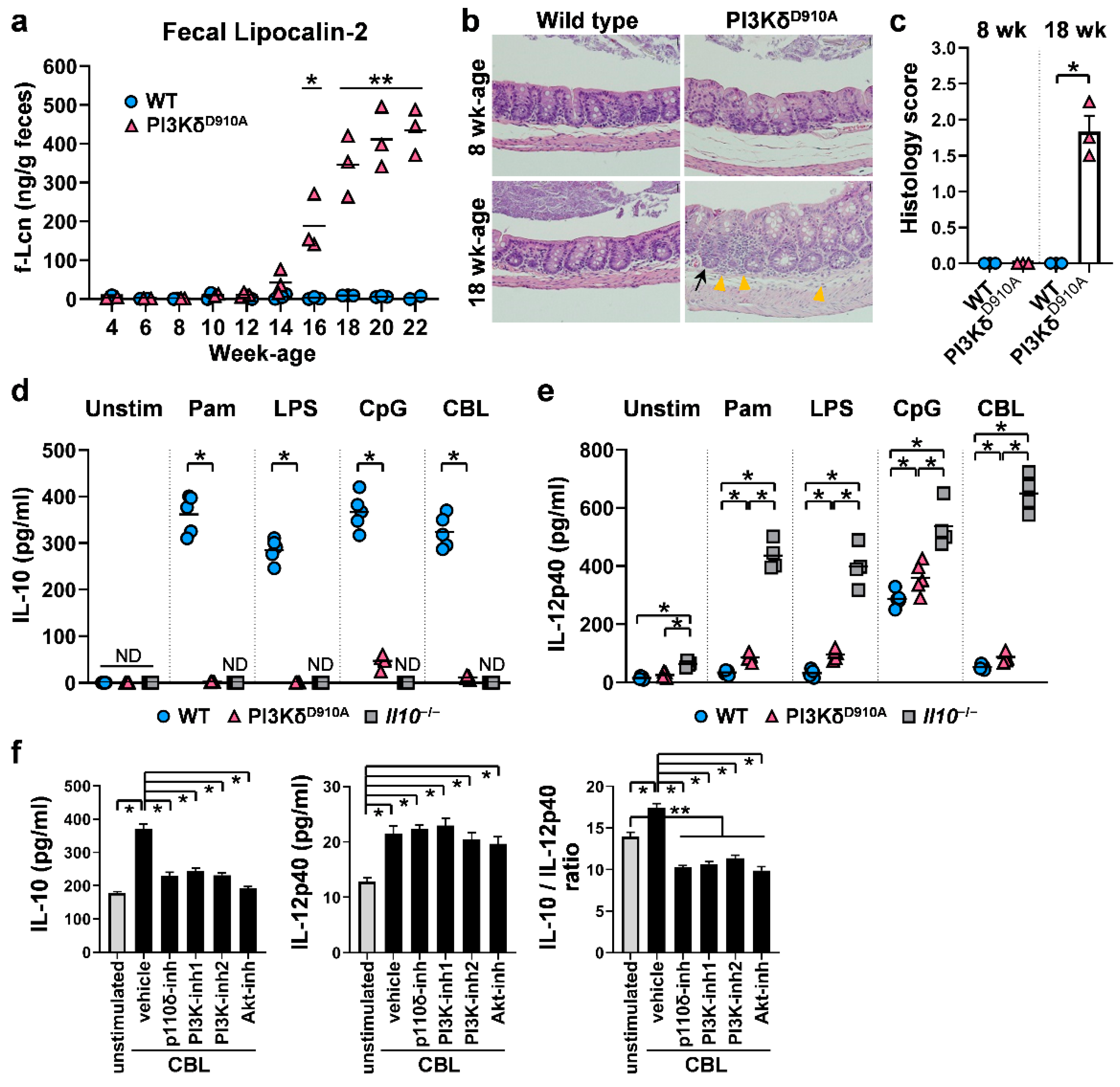
3.1. Inflammatory (IL-12p40) or Regulatory (IL-10) Pathways are Determined by PI3Kδ Signaling When Stimulated by Resident Bacteria
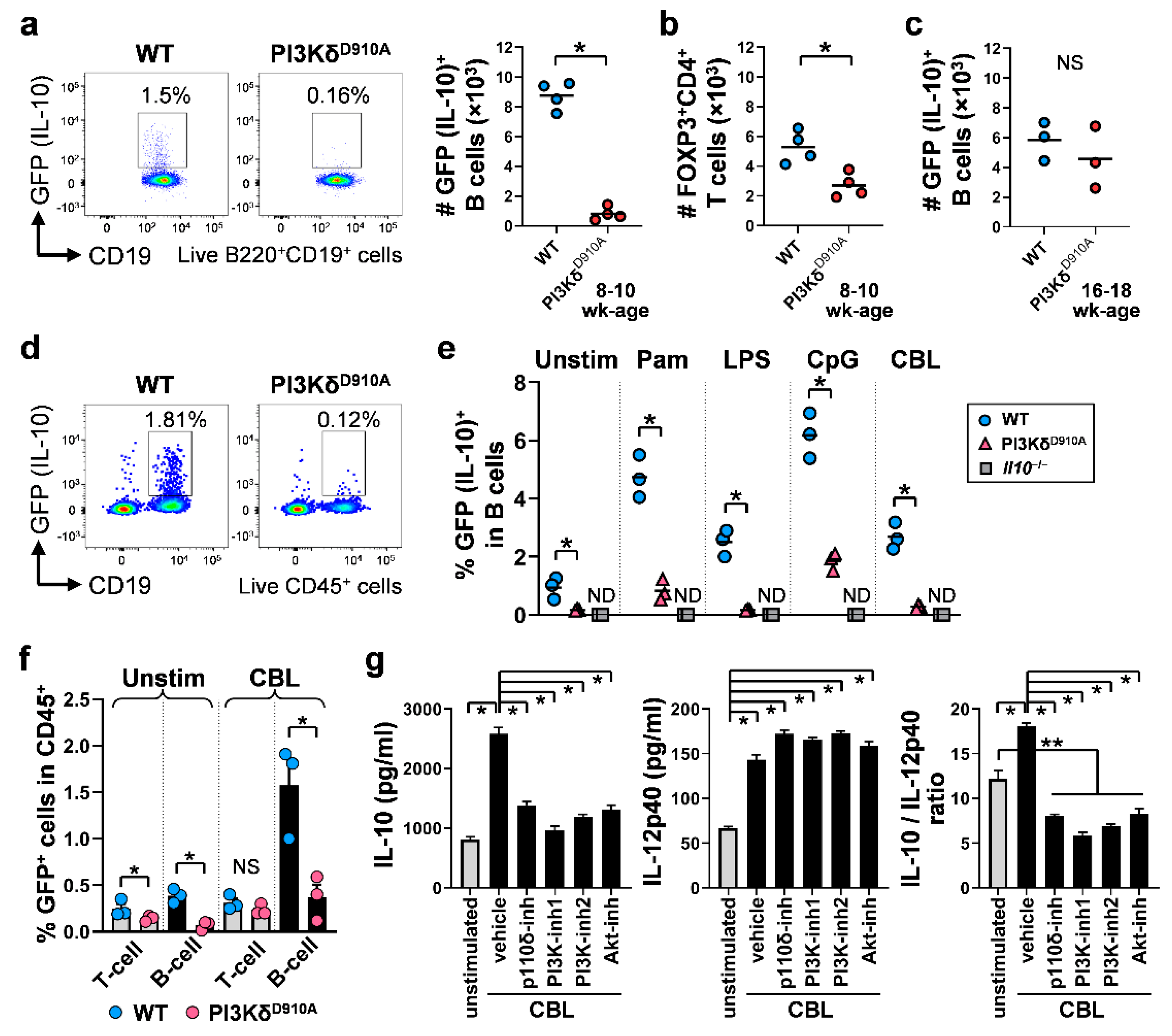
3.2. Colonic IL-10-Producing B Cells Are Decreased in PI3KδD910A Mice
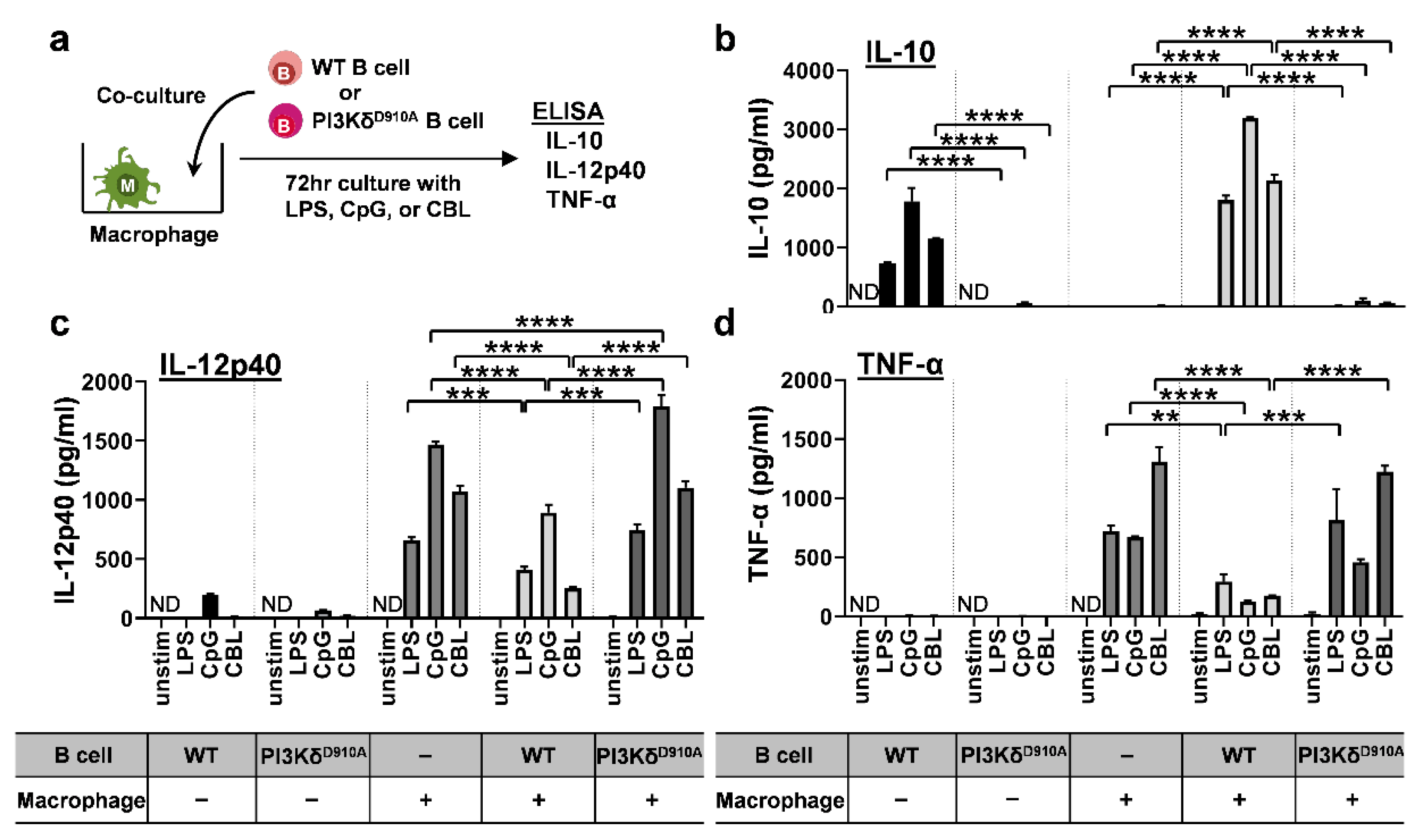
3.3. Bacteria-Stimulated PI3KδD910A B Cells Do Not Suppress Pro-Inflammatory Cytokine Secretion by Macrophages
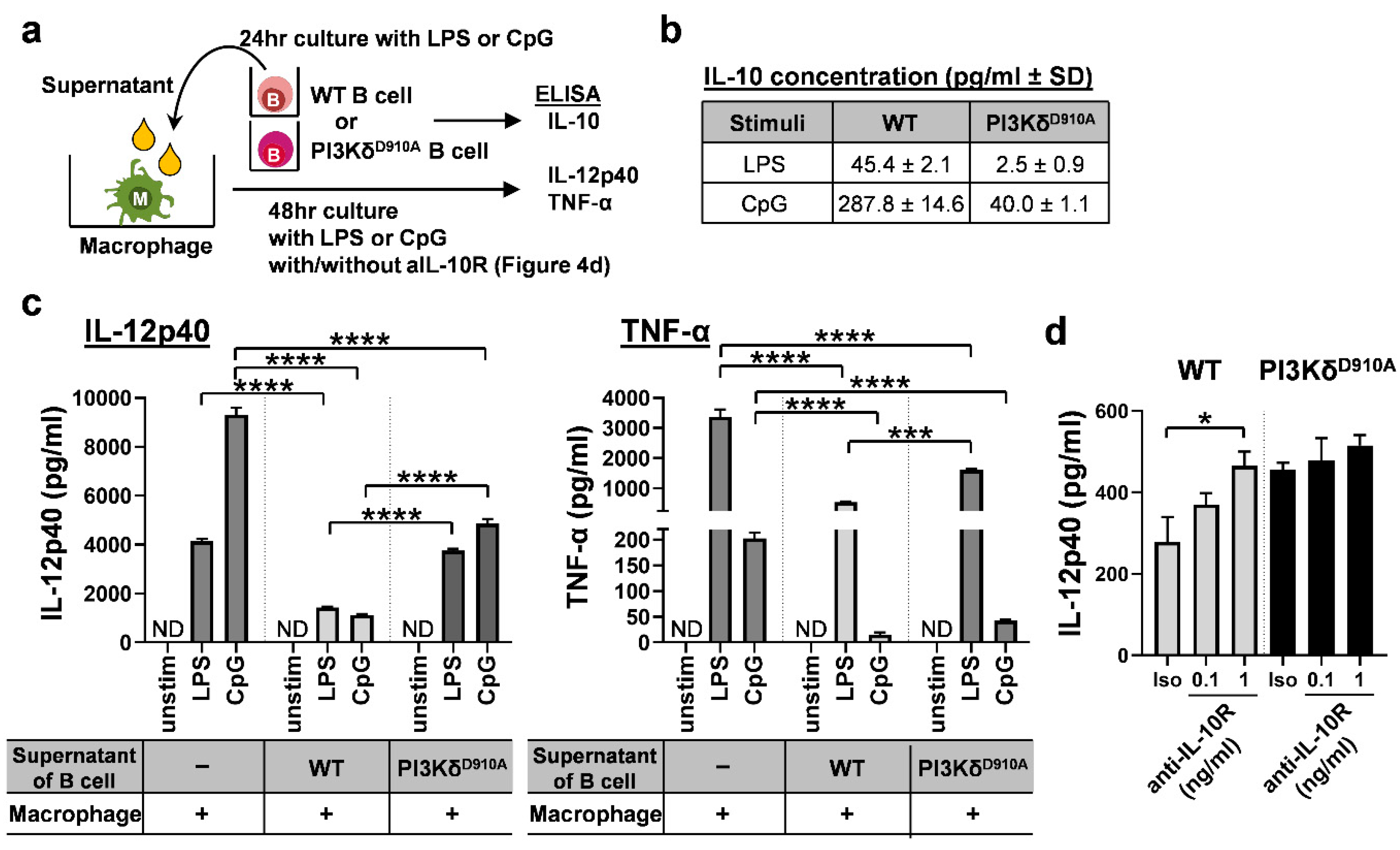
3.4. Secreted IL-10 is the Dominant Way B Cells Regulate Pro-Inflammatory Cytokine Production in Macrophages in Response to Bacterial Products
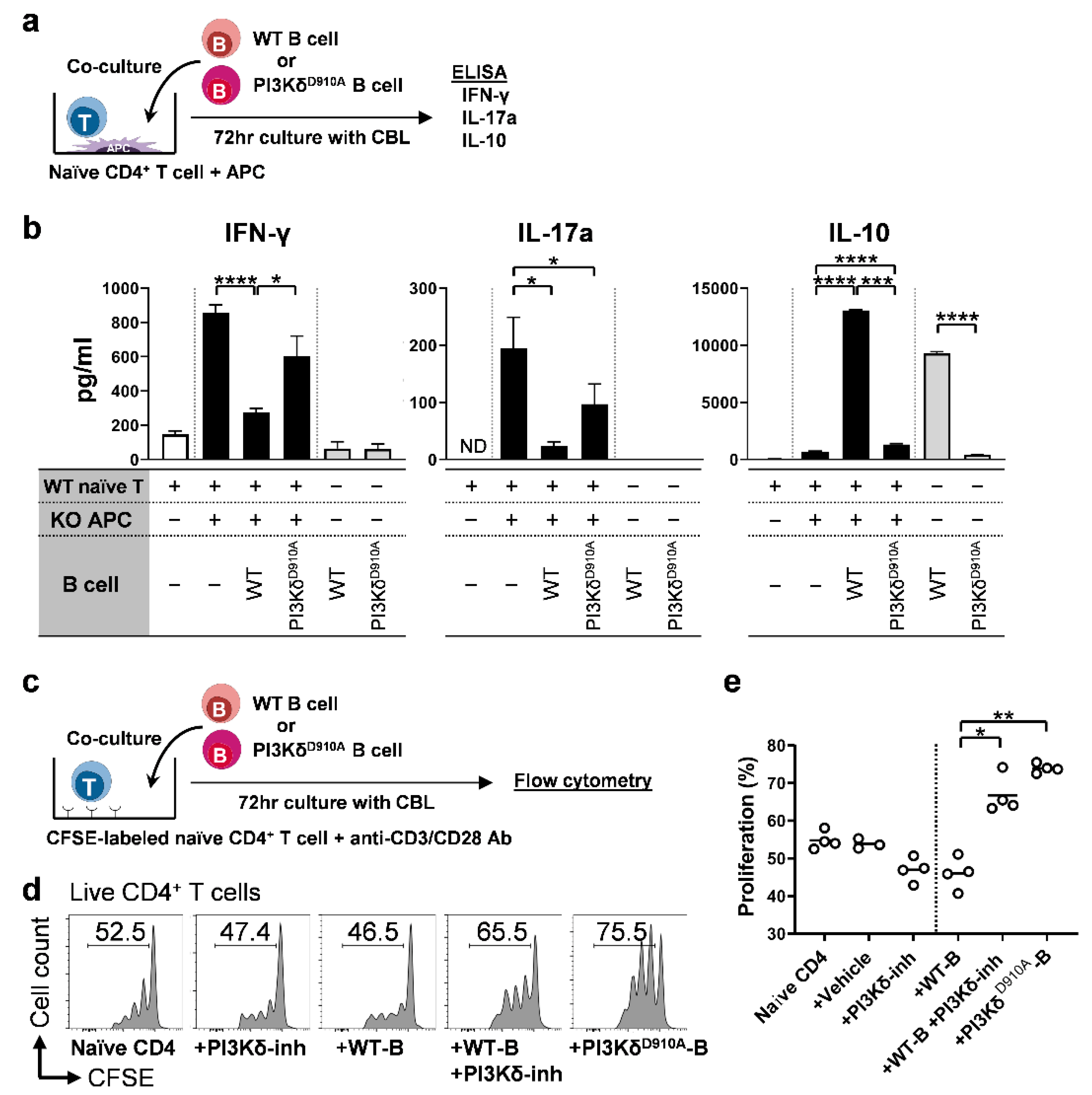
3.5. PI3Kδ-Signaling in B Cells Regulates Bacterial-Induced Pro-Inflammatory Cytokine Secretion by T Cells
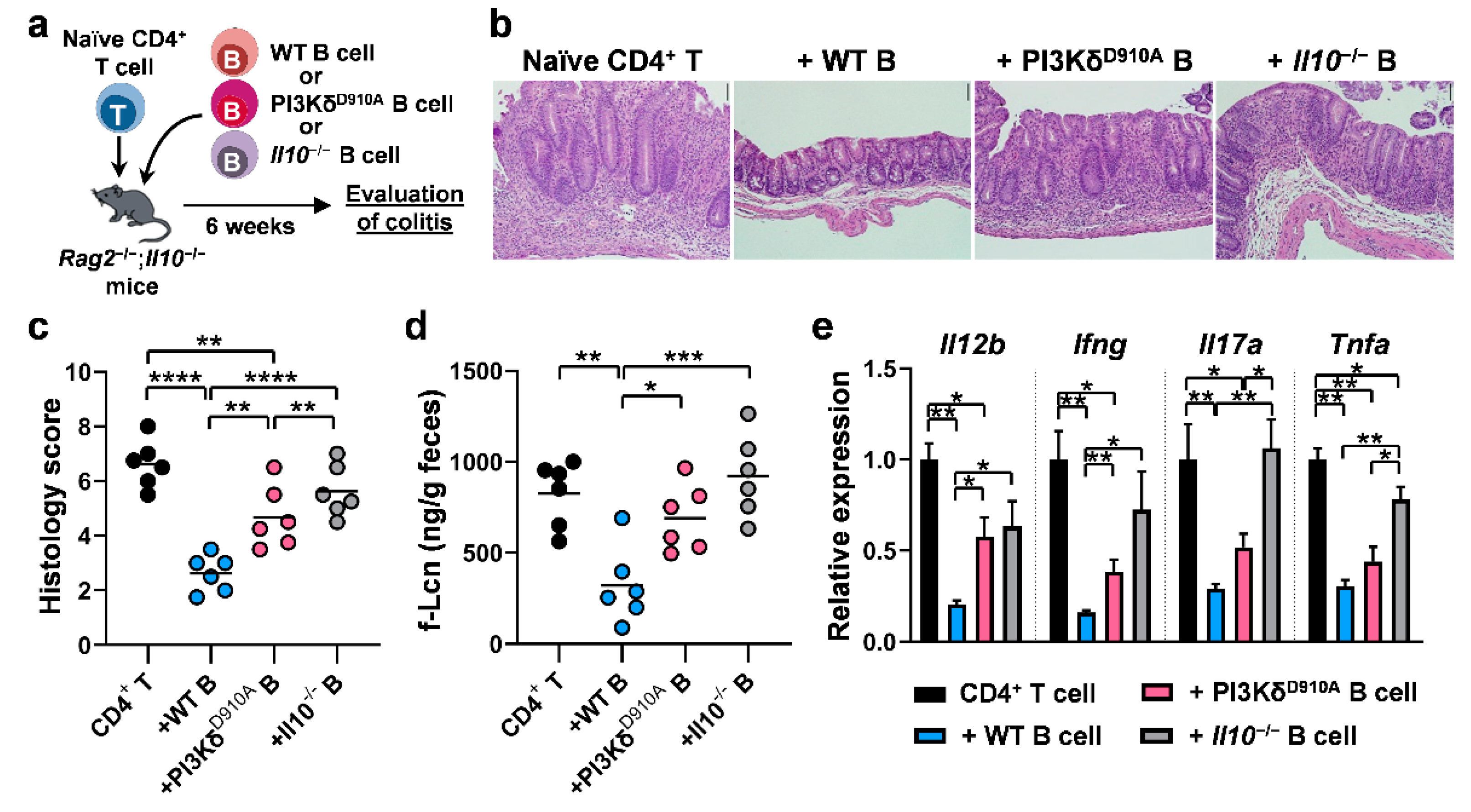
3.6. PI3Kδ-Signaling in B Cells is Required to Confer Protection Against T Cell-Mediated Colitis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, M.; Pohin, M.; Powrie, F. Cytokine Networks in the Pathophysiology of Inflammatory Bowel Disease. Immunity 2019, 50, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.-C.; Montgomery, S.A.; Truax, A.D.; June Brickey, W.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 1–14. [Google Scholar]
- Atarashi, K.; Honda, K. Microbiota in autoimmunity and tolerance. Curr. Opin. Immunol. 2011, 23, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Oka, A.; Liu, B.; Herzog, J.W.; Eun, C.S.; Fan, T.-J.; Bulik-Sullivan, E.; Carroll, I.M.; Hansen, J.J.; Chen, L.; et al. Microbiota maintain colonic homeostasis by activating TLR2/MyD88/PI3K signaling in IL-10-producing regulatory B cells. J. Clin. Investig. 2019, 129, 130. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Littman, D.R. Modulation of immune homeostasis by commensal bacteria. Curr. Opin. Microbiol. 2011, 14, 106–114. [Google Scholar] [CrossRef] [Green Version]
- Makita, S.; Kanai, T.; Oshima, S.; Uraushihara, K.; Totsuka, T.; Sawada, T.; Nakamura, T.; Koganei, K.; Fukushima, T.; Watanabe, M. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J. Immunol. 2004, 173, 3119–3130. [Google Scholar] [CrossRef]
- Mishima, Y.; Ishihara, S.; Aziz, M.M.; Oka, A.; Kusunoki, R.; Otani, A.; Tada, Y.; Li, Y.Y.; Moriyama, I.; Oshima, N.; et al. Decreased production of interleukin-10 and transforming growth factor-β in Toll-like receptor-activated intestinal B cells in SAMP1/Yit mice. Immunology 2010, 131, 473–487. [Google Scholar] [CrossRef]
- Oka, A.; Ishihara, S.; Mishima, Y.; Tada, Y.; Kusunoki, R.; Fukuba, N.; Yuki, T.; Kawashima, K.; Matsumoto, S.; Kinoshita, Y. Role of Regulatory B Cells in Chronic Intestinal Inflammation: Association with Pathogenesis of Crohn’s Disease. Inflamm. Bowel Dis. 2014, 20, 315–328. [Google Scholar] [CrossRef]
- Mishima, Y.; Liu, B.; Hansen, J.J.; Sartor, R.B. Resident bacteria-stimulated IL-10-secreting B cells ameliorate T cell-mediated colitis by inducing Tr-1 cells that require IL-27-signaling. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Uno, J.K.; Rao, K.N.; Matsuoka, K.; Sheikh, S.Z.; Kobayashi, T.; Li, F.; Steinbach, E.C.; Sepulveda, A.R.; Vanhaesebroeck, B.; Sartor, R.B.; et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110δ. Gastroenterology 2010, 139, 1642–1653.e6. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, E.J.; Matzinger, P. B cells turn off virgin but not memory T cells. Science 1992, 258, 1156–1159. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, A.; Mizoguchi, E.; Takedatsu, H.; Blumberg, R.S.; Bhan, A.K. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity 2002, 16, 219–230. [Google Scholar] [CrossRef]
- El Fassi, D.; Nielsen, C.H.; Kjeldsen, J.; Clemmensen, O.; Hegedüs, L. Ulcerative colitis following B lymphocyte depletion with rituximab in a patient with Graves’ disease. Gut 2008, 57, 714–715. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.; Atreya, R.; Ghalibafian, M.; Galle, P.R.; Neurath, M.F. Exacerbation of ulcerative colitis after rituximab salvage therapy. Inflamm. Bowel Dis. 2007, 13, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Yanaba, K.; Yoshizaki, A.; Asano, Y.; Kadono, T.; Tedder, T.F.; Sato, S. IL-10-producing regulatory B10 cells inhibit intestinal injury in a mouse model. Am. J. Pathol. 2011, 178, 735–743. [Google Scholar] [CrossRef]
- Matsushita, T.; Le Huu, D.; Kobayashi, T.; Hamaguchi, Y.; Hasegawa, M.; Naka, K.; Hirao, A.; Muramatsu, M.; Takehara, K.; Fujimoto, M. A novel splenic B1 regulatory cell subset suppresses allergic disease through phosphatidylinositol 3-kinase-Akt pathway activation. J. Allergy Clin. Immunol. 2016, 138, 1170–1182.e9. [Google Scholar] [CrossRef]
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem. Sci. 2009, 34, 115–127. [Google Scholar] [CrossRef]
- Kuo, C.-C.; Lin, W.-T.; Liang, C.-M.; Liang, S.-M. Class I and III phosphatidylinositol 3’-kinase play distinct roles in TLR signaling pathway. J. Immunol. 2006, 176, 5943–5949. [Google Scholar] [CrossRef]
- Fukao, T.; Koyasu, S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Liew, F.Y.; Xu, D.; Brint, E.K.; O’Neill, L.A.J. Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005, 5, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Cahill, C.M.; Rogers, J.T.; Walker, W.A. The role of phosphoinositide 3-kinase signaling in intestinal inflammation. J. Signal Transduct. 2012, 2012, 358476. [Google Scholar] [CrossRef] [PubMed]
- Fukao, T.; Tanabe, M.; Terauchi, Y.; Ota, T.; Matsuda, S.; Asano, T.; Kadowaki, T.; Takeuchi, T.; Koyasu, S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 2002, 3, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Schifferle, R.E.; Cuesta, N.; Vogel, S.N.; Katz, J.; Michalek, S.M. Role of the phosphatidylinositol 3 kinase-Akt pathway in the regulation of IL-10 and IL-12 by Porphyromonas gingivalis lipopolysaccharide. J. Immunol. 2003, 171, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Garcia, C.A.; Rehani, K.; Cekic, C.; Alard, P.; Kinane, D.F.; Mitchell, T.; Martin, M. IFN-beta production by TLR4-stimulated innate immune cells is negatively regulated by GSK3-beta. J. Immunol. 2008, 181, 6797–6802. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef]
- Seki, N.; Nimura, Y.; Ohira, M.; Saito, T.; Ichimiya, S.; Nomura, N.; Nakagawara, A. Identification and chromosome assignment of a human gene encoding a novel phosphatidylinositol-3 kinase. DNA Res. 1997, 4, 355–358. [Google Scholar] [CrossRef]
- Cho, J.H.; Nicolae, D.L.; Ramos, R.; Fields, C.T.; Rabenau, K.; Corradino, S.; Brant, S.R.; Espinosa, R.; LeBeau, M.; Hanauer, S.B.; et al. Linkage and linkage disequilibrium in chromosome band 1p36 in American Chaldeans with inflammatory bowel disease. Hum. Mol. Genet. 2000, 9, 1425–1432. [Google Scholar] [CrossRef] [Green Version]
- Conley, M.E.; Dobbs, A.K.; Quintana, A.M.; Bosompem, A.; Wang, Y.-D.; Coustan-Smith, E.; Smith, A.M.; Perez, E.E.; Murray, P.J. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J. Exp. Med. 2012, 209, 463–470. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, E.C.; Kobayashi, T.; Russo, S.M.; Sheikh, S.Z.; Gipson, G.R.; Kennedy, S.T.; Uno, J.K.; Mishima, Y.; Borst, L.B.; Liu, B.; et al. Innate PI3K p110δ regulates Th1/Th17 development and microbiota-dependent colitis. J. Immunol. 2014, 192, 3958–3968. [Google Scholar] [CrossRef] [PubMed]
- Patton, D.T.; Garden, O.A.; Pearce, W.P.; Clough, L.E.; Monk, C.R.; Leung, E.; Rowan, W.C.; Sancho, S.; Walker, L.S.K.; Vanhaesebroeck, B.; et al. Cutting edge: The phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Immunol. 2006, 177, 6598–6602. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.; Demircik, F.; Surianarayanan, S.; Allen, J.L.; Divanovic, S.; Trompette, A.; Yogev, N.; Gu, Y.; Khodoun, M.; Hildeman, D.; et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J. Immunol. 2009, 183, 2312–2320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Tonkonogy, S.L.; Albright, C.A.; Tsang, J.; Balish, E.J.; Braun, J.; Huycke, M.M.; Sartor, R.B. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 2005, 128, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Tonkonogy, S.L.; Sartor, R.B. Antigen-presenting cell production of IL-10 inhibits T-helper 1 and 17 cell responses and suppresses colitis in mice. Gastroenterology 2011, 141, 653–662.e4. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Hold, G.L.; Harmsen, H.J.M.; Stewart, C.S.; Flint, H.J. Growth requirements and fermentation products of Fusobacterium prausnitzii, and a proposal to reclassify it as Faecalibacterium prausnitzii gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 2141–2146. [Google Scholar] [Green Version]
- Chassaing, B.; Srinivasan, G.; Delgado, M.A.; Young, A.N.; Gewirtz, A.T.; Vijay-Kumar, M. Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLoS ONE 2012, 7, e44328. [Google Scholar] [CrossRef]
- Scapini, P.; Lamagna, C.; Hu, Y.; Lee, K.; Tang, Q.; DeFranco, A.L.; Lowell, C.A. B cell-derived IL-10 suppresses inflammatory disease in Lyn-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, E823–E832. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.; Noh, G.; Lee, S.J.; Lee, J.H.; Kim, A.; Kim, H.S.; Choi, W.S. Tolerogenic effects of interferon-gamma with induction of allergen-specific interleukin-10-producing regulatory B cell (Br1) changes in non-IgE-mediated food allergy. Cell. Immunol. 2012, 273, 140–149. [Google Scholar] [CrossRef]
- Van der Veeken, J.; Gonzalez, A.J.; Cho, H.; Arvey, A.; Hemmers, S.; Leslie, C.S.; Rudensky, A.Y. Memory of Inflammation in Regulatory T Cells. Cell 2016, 166, 977–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramadani, F.; Bolland, D.J.; Garcon, F.; Emery, J.L.; Vanhaesebroeck, B.; Corcoran, A.E.; Okkenhaug, K. The PI3K isoforms p110alpha and p110delta are essential for pre-B cell receptor signaling and B cell development. Sci. Signal. 2010, 3, ra60. [Google Scholar] [CrossRef] [PubMed]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [PubMed]
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta 2015, 1851, 882–897. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, A.; Bhan, A.K. A case for regulatory B cells. J. Immunol. 2006, 176, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Tedder, T.F. B10 Cells: A Functionally Defined Regulatory B Cell Subset. J. Immunol. 2015, 194, 1395–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, A.; Lau, C.W.; Mizoguchi, E.; Mizoguchi, A. Regulatory B cells in mouse models of intestinal inflammation. Methods Mol. Biol. 2014, 1190, 227–241. [Google Scholar] [PubMed]
- Coutré, S.E.; Barrientos, J.C.; Brown, J.R.; de Vos, S.; Furman, R.R.; Keating, M.J.; Li, D.; O’Brien, S.M.; Pagel, J.M.; Poleski, M.H.; et al. Management of adverse events associated with idelalisib treatment: Expert panel opinion. Leuk. Lymphoma 2015, 56, 2779–2786. [Google Scholar] [CrossRef]
- Randis, T.M.; Puri, K.D.; Zhou, H.; Diacovo, T.G. Role of PI3Kdelta and PI3Kgamma in inflammatory arthritis and tissue localization of neutrophils. Eur. J. Immunol. 2008, 38, 1215–1224. [Google Scholar] [CrossRef]
- Lee, K.S.; Lee, H.K.; Hayflick, J.S.; Lee, Y.C.; Puri, K.D. Inhibition of phosphoinositide 3-kinase delta attenuates allergic airway inflammation and hyperresponsiveness in murine asthma model. FASEB J. 2006, 20, 455–465. [Google Scholar] [CrossRef]
- Doukas, J.; Eide, L.; Stebbins, K.; Racanelli-Layton, A.; Dellamary, L.; Martin, M.; Dneprovskaia, E.; Noronha, G.; Soll, R.; Wrasidlo, W.; et al. Aerosolized phosphoinositide 3-kinase gamma/delta inhibitor TG100-115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol] as a therapeutic candidate for asthma and chronic obstructive pulmonary disease. J. Pharmacol. Exp. Ther. 2009, 328, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Nashed, B.F.; Zhang, T.; Al-Alwan, M.; Srinivasan, G.; Halayko, A.J.; Okkenhaug, K.; Vanhaesebroeck, B.; Hayglass, K.T.; Marshall, A.J. Role of the phosphoinositide 3-kinase p110delta in generation of type 2 cytokine responses and allergic airway inflammation. Eur. J. Immunol. 2007, 37, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Ali, K.; Camps, M.; Pearce, W.P.; Ji, H.; Rückle, T.; Kuehn, N.; Pasquali, C.; Chabert, C.; Rommel, C.; Vanhaesebroeck, B. Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J. Immunol. 2008, 180, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Haylock-Jacobs, S.; Comerford, I.; Bunting, M.; Kara, E.; Townley, S.; Klingler-Hoffmann, M.; Vanhaesebroeck, B.; Puri, K.D.; McColl, S.R. PI3Kδ drives the pathogenesis of experimental autoimmune encephalomyelitis by inhibiting effector T cell apoptosis and promoting Th17 differentiation. J. Autoimmun. 2011, 36, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Roller, A.; Perino, A.; Dapavo, P.; Soro, E.; Okkenhaug, K.; Hirsch, E.; Ji, H. Blockade of phosphatidylinositol 3-kinase PI3Kδ or PI3Kγ reduces IL-17 and ameliorates imiquimod-induced psoriasis-like dermatitis. J. Immunol. 2012, 189, 4612–4620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Wei, P.; Zhang, H.; Liu, C. Inhibition of PI3Kδ improves systemic lupus in mice. Inflammation 2014, 37, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Chellappa, S.; Kushekhar, K.; Munthe, L.A.; Tjønnfjord, G.E.; Aandahl, E.M.; Okkenhaug, K.; Taskén, K. The PI3K p110δ Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J. Immunol. 2019, 202, 1397–1405. [Google Scholar] [CrossRef]
- Coulter, T.I.; Chandra, A.; Bacon, C.M.; Babar, J.; Curtis, J.; Screaton, N.; Goodlad, J.R.; Farmer, G.; Steele, C.L.; Leahy, T.R.; et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 2017, 139, 597–606.e4. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Patton, D.T.; Bilancio, A.; Garçon, F.; Rowan, W.C.; Vanhaesebroeck, B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J. Immunol. 2006, 177, 5122–5128. [Google Scholar] [CrossRef]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef]
- Kucharzik, T.; Stoll, R.; Lügering, N.; Domschke, W. Circulating antiinflammatory cytokine IL-10 in patients with inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 100, 452–456. [Google Scholar] [CrossRef]
- Cope, A.; Le Friec, G.; Cardone, J.; Kemper, C. The Th1 life cycle: Molecular control of IFN-γ to IL-10 switching. Trends Immunol. 2011, 32, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Meyaard, L.; Hovenkamp, E.; Otto, S.A.; Miedema, F. IL-12-induced IL-10 production by human T cells as a negative feedback for IL-12-induced immune responses. J. Immunol. 1996, 156, 2776–2782. [Google Scholar] [PubMed]
- Dambuza, I.M.; He, C.; Choi, J.K.; Yu, C.-R.; Wang, R.; Mattapallil, M.J.; Wingfield, P.T.; Caspi, R.R.; Egwuagu, C.E. IL-12p35 induces expansion of IL-10 and IL-35-expressing regulatory B cells and ameliorates autoimmune disease. Nat. Commun. 2017, 8, 719. [Google Scholar] [CrossRef] [PubMed]
- Fadlallah, J.; Kafsi, H.E.; Sterlin, D.; Juste, C.; Parizot, C.; Dorgham, K.; Autaa, G.; Gouas, D.; Almeida, M.; Lepage, P.; et al. Microbial ecology perturbation in human IgA deficiency. Sci. Transl. Med. 2018, 10, eaan1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madsen, K.L.; Doyle, J.S.; Jewell, L.D.; Tavernini, M.M.; Fedorak, R.N. Lactobacillus species prevents colitis in interleukin 10 gene-deficient mice. Gastroenterology 1999, 116, 1107–1114. [Google Scholar] [CrossRef]
- Burnham, C.-A.D.; Shokoples, S.E.; Tyrrell, G.J. Invasion of HeLa cells by group B streptococcus requires the phosphoinositide-3-kinase signalling pathway and modulates phosphorylation of host-cell Akt and glycogen synthase kinase-3. Microbiology 2007, 153, 4240–4252. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oka, A.; Mishima, Y.; Liu, B.; Herzog, J.W.; Steinbach, E.C.; Kobayashi, T.; Plevy, S.E.; Sartor, R.B. Phosphoinositide 3-Kinase P110δ-Signaling Is Critical for Microbiota-Activated IL-10 Production by B Cells that Regulate Intestinal Inflammation. Cells 2019, 8, 1121. https://doi.org/10.3390/cells8101121
Oka A, Mishima Y, Liu B, Herzog JW, Steinbach EC, Kobayashi T, Plevy SE, Sartor RB. Phosphoinositide 3-Kinase P110δ-Signaling Is Critical for Microbiota-Activated IL-10 Production by B Cells that Regulate Intestinal Inflammation. Cells. 2019; 8(10):1121. https://doi.org/10.3390/cells8101121
Chicago/Turabian StyleOka, Akihiko, Yoshiyuki Mishima, Bo Liu, Jeremy W. Herzog, Erin C. Steinbach, Taku Kobayashi, Scott E. Plevy, and R. Balfour Sartor. 2019. "Phosphoinositide 3-Kinase P110δ-Signaling Is Critical for Microbiota-Activated IL-10 Production by B Cells that Regulate Intestinal Inflammation" Cells 8, no. 10: 1121. https://doi.org/10.3390/cells8101121
APA StyleOka, A., Mishima, Y., Liu, B., Herzog, J. W., Steinbach, E. C., Kobayashi, T., Plevy, S. E., & Sartor, R. B. (2019). Phosphoinositide 3-Kinase P110δ-Signaling Is Critical for Microbiota-Activated IL-10 Production by B Cells that Regulate Intestinal Inflammation. Cells, 8(10), 1121. https://doi.org/10.3390/cells8101121