A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking



Abstract
:1. Introduction
2. Material and Methods
2.1. Preparation of Compound List and Calculation of Chemical Descriptors
2.2. P-Glycoprotein Modulator Prediction Model Establishment
2.3. Molecular Docking
- Ki (M)
- ΔG (cal/mol) = 1000 * LBE (lowest binding energy, kcal/mol)
- R (cal/mol-K): gas constant, 1.986 cal/mol-K
- T (K): room temperature, 298 K
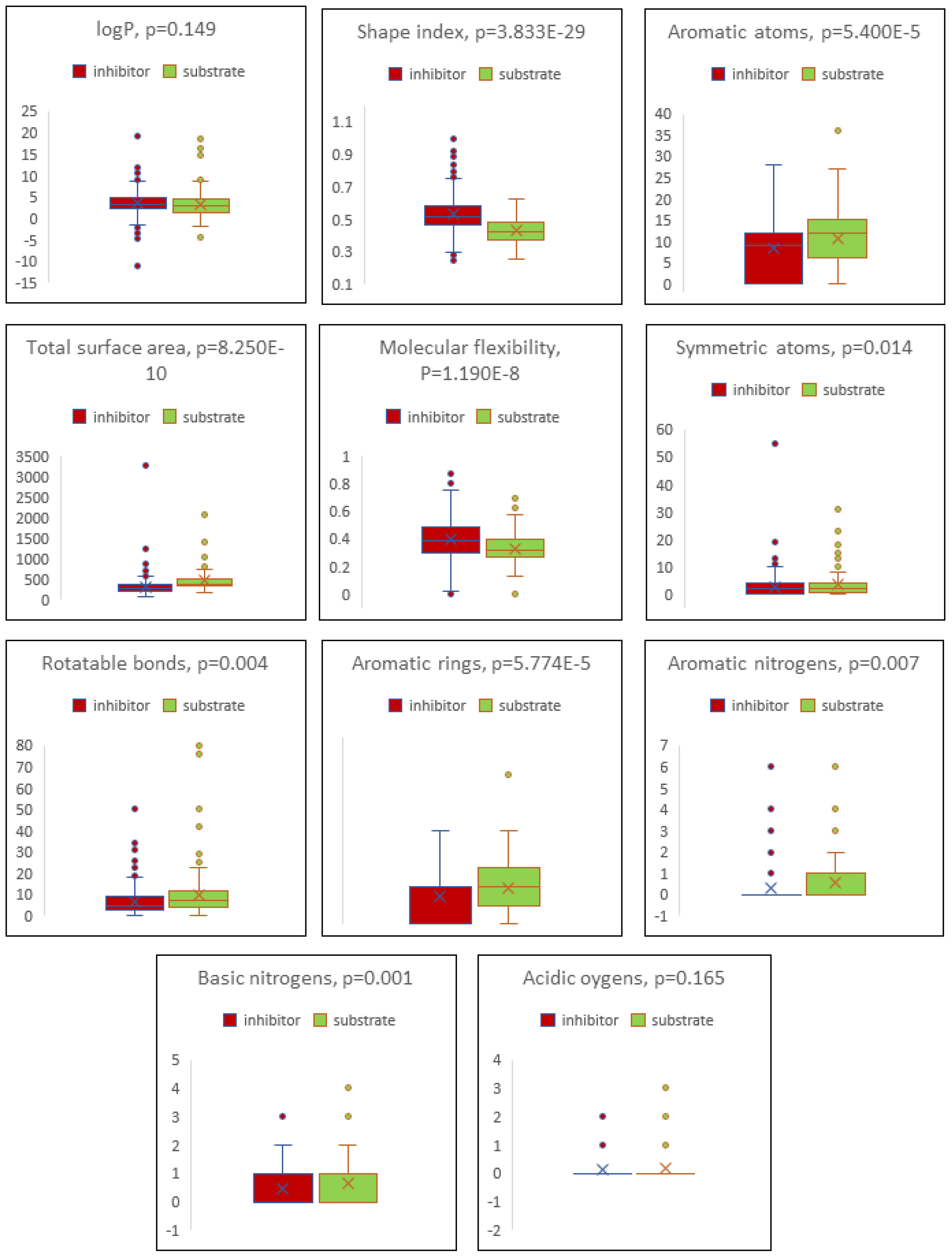
2.4. Boxplot Analysis
3. Results
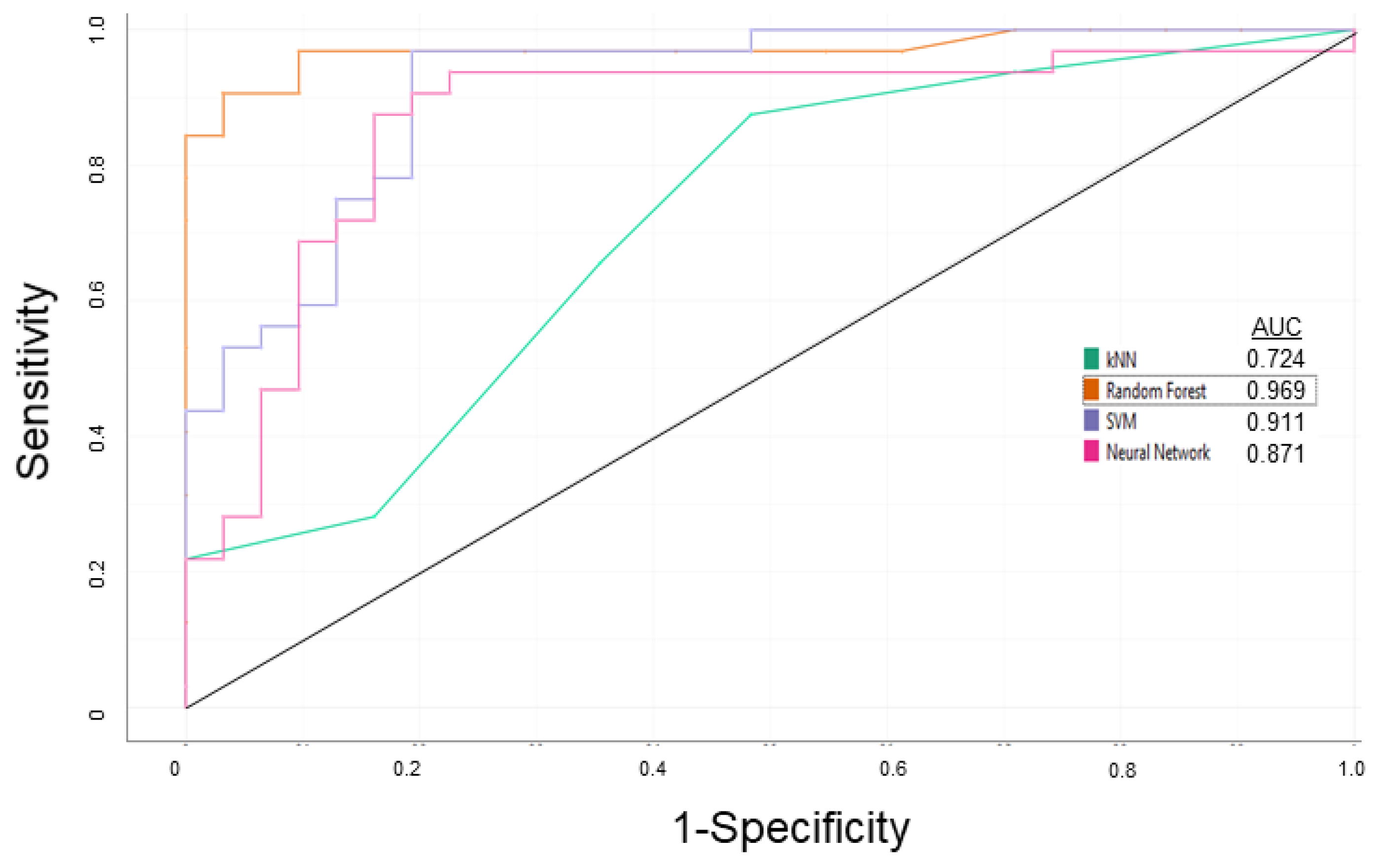
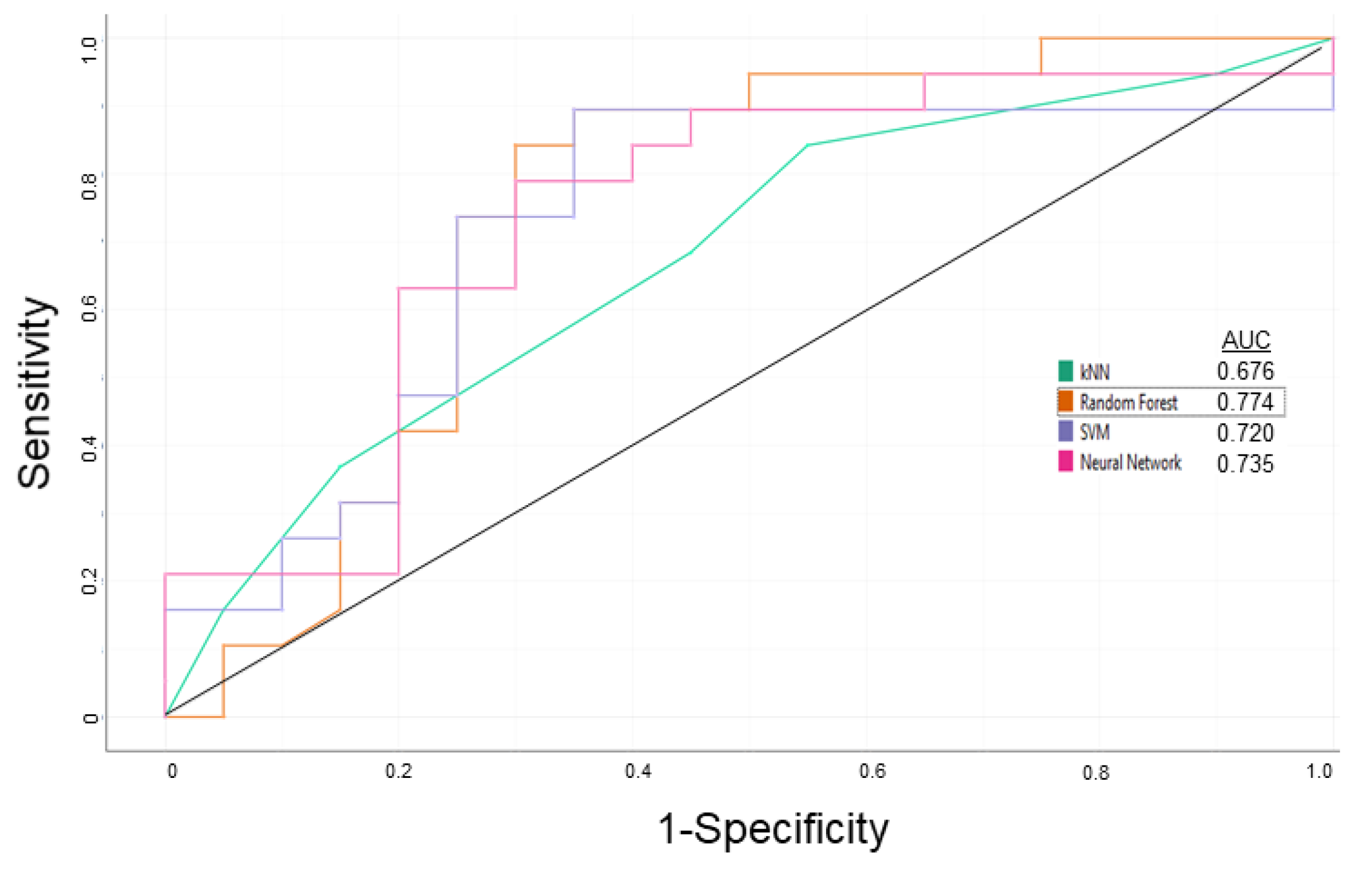
3.1. P-glycoprotein Modulator Predictions
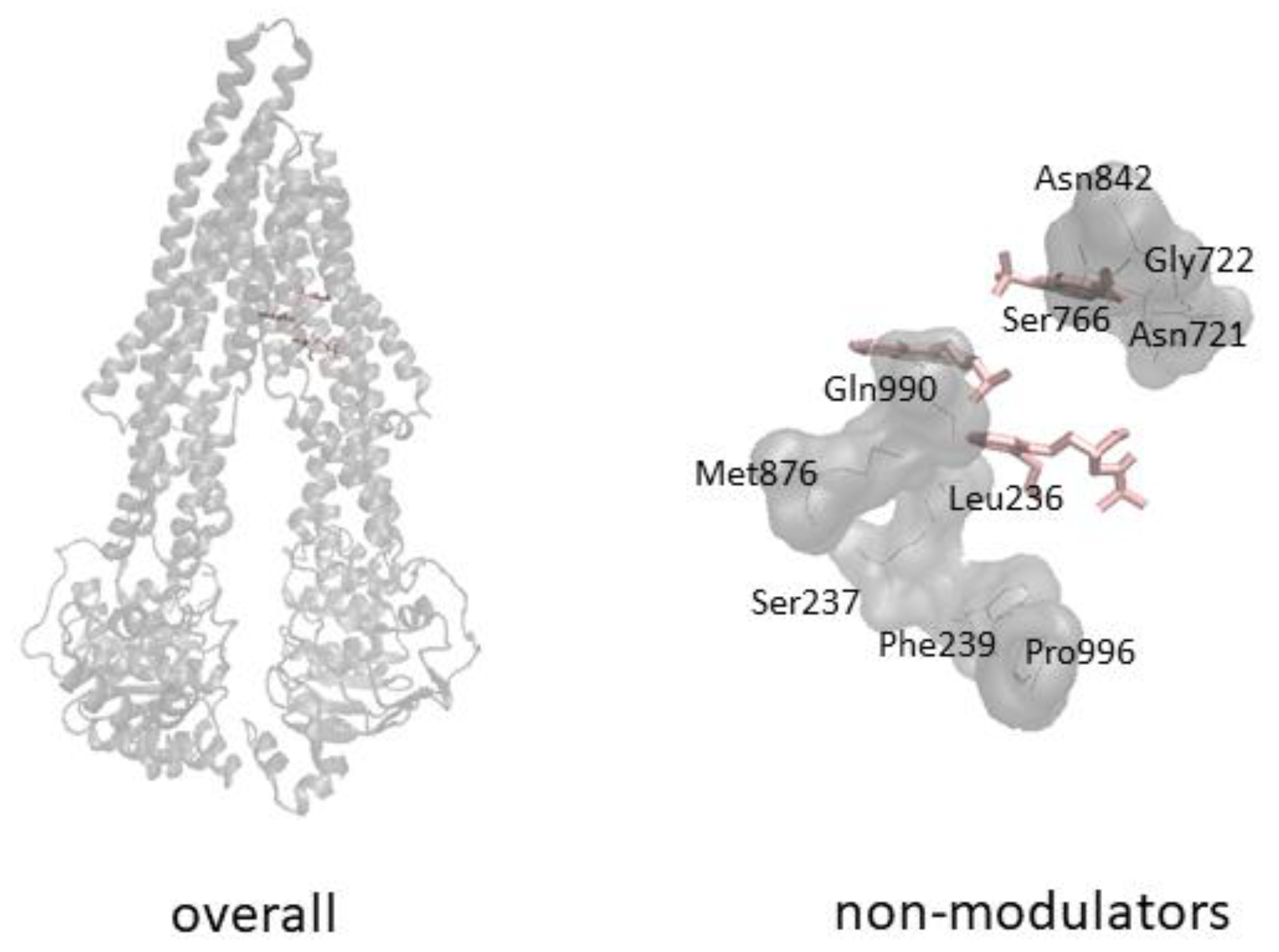
3.2. Molecular Docking
4. Discussion
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| AUC | area under the curve |
| kNN | k-nearest neighboring |
| MDR | multidrug resistance |
| P-gp | P-glycoprotein |
| RF | random forest |
| ROC | receiver operating characteristic |
| SVM | support vector machine |
References
- Efferth, T.; Zeino, M.; Volm, M. Modulation of P-glycoprotein-mediated multidrug resistance by synthetic and phytochemical small molecules, monoclonal antibodies, and therapeutic nucleic acids. In Resistance to Targeted ABC Transporters in Cancer; Efferth, T., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 153–181. [Google Scholar] [CrossRef]
- Krech, T.; Scheuerer, E.; Geffers, R.; Kreipe, H.; Lehmann, U.; Christgen, M. ABCB1/MDR1 contributes to the anticancer drug-resistant phenotype of IPH-926 human lobular breast cancer cells. Cancer Lett. 2012, 315, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; Nooter, K. Pharmacokinetic resistance to imatinib mesylate—Role of the ABC drug pumps ABCG2 (BCRP) and ABCB1 (MDR1) in the oral bioavailability of imatinib. Cell Cycle 2004, 3, 1502–1505. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. Inhibition of P-glycoprotein at the blood brain barrier by phytochemicals derived from traditional Chinese medicine. Planta Medica 2009, 75, SL3. [Google Scholar] [CrossRef]
- Kuete, V.; Fouotsa, H.; Mbaveng, A.T.; Wiench, B.; Nkengfack, A.E.; Efferth, T. Cytotoxicity of a naturally occurring furoquinoline alkaloid and four acridone alkaloids towards multi-factorial drug-resistant cancer cells. Phytomedicine 2015, 22, 946–951. [Google Scholar] [CrossRef] [PubMed]
- Kadioglu, O.; Cao, J.; Kosyakova, N.; Mrasek, K.; Liehr, T.; Efferth, T. Genomic and transcriptomic profiling of resistant CEM/ADR-5000 and sensitive CCRF-CEM leukaemia cells for unravelling the full complexity of multi-factorial multidrug resistance. Sci. Rep. 2016, 6, 36754. [Google Scholar] [CrossRef]
- Kuete, V.; Saeed, M.E.M.; Kadioglu, O.; Bortzler, J.; Khalid, H.; Greten, H.J.; Efferth, T. Pharmacogenomic and molecular docking studies on the cytotoxicity of the natural steroid wortmannin against multidrug-resistant tumor cells. Phytomedicine 2015, 22, 120–127. [Google Scholar] [CrossRef]
- Efferth, T.; Osieka, R. Clinical Relevance of the Mdr-1 gene and its gene-product, P-glycoprotein for cancer chemotherapy—a metaanalysis. Tumordiagn Ther. 1993, 14, 238–243. [Google Scholar]
- Efferth, T. The human ATP-binding cassette transporter genes: From the bench to the bedside. Curr. Mol. Med. 2001, 1, 45–65. [Google Scholar] [CrossRef]
- Gillet, J.P.; Efferth, T.; Remacle, J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim. Biophys. Acta Rev. Cancer 2007, 1775, 237–262. [Google Scholar] [CrossRef]
- Kadioglu, O.; Saeed, M.E.M.; Valoti, M.; Frosini, M.; Sgaragli, G.; Efferth, T. Interactions of human P-glycoprotein transport substrates and inhibitors at the drug binding domain: Functional and molecular docking analyses. Biochem. Pharmacol. 2016, 104, 42–51. [Google Scholar] [CrossRef]
- Srinivas, N.R. Understanding the role of tariquidar, a potent Pgp inhibitor, in combination trials with cytotoxic drugs: What is missing? Cancer Chemother. Pharmacol. 2016, 78, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.; Edgerly, M.; Wilson, R.; Chen, C.; Rutt, A.; Bakke, S.; Robey, R.; Dwyer, A.; Goldspiel, B.; Balis, F.; et al. A phase I study of the P-glycoprotein antagonist tariquidar in combination with vinorelbine. Clin. Cancer Res. 2009, 15, 3574–3582. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.; Widemann, B.C.; Pastakia, D.; Chen, C.C.; Yang, S.X.; Cole, D.; Balis, F.M. Pharmacokinetic and pharmacodynamic study of tariquidar (XR9576), a P-glycoprotein inhibitor, in combination with doxorubicin, vinorelbine, or docetaxel in children and adolescents with refractory solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.M.; Wang, Y.C.; Zhou, W.N.; Fan, Y.R.; Zhao, J.N.; Zhu, L.; Lu, S.; Lu, T.; Chen, Y.D.; Liu, H.C. A combined drug discovery strategy based on machine learning and molecular docking. Chem. Boil. Drug Des. 2019, 93, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Zoffmann, S.; Vercruysse, M.; Benmansour, F.; Maunz, A.; Wolf, L.; Marti, R.B.; Heckel, T.; Ding, H.Y.; Truong, H.H.; Prummer, M.; et al. Machine learning-powered antibiotics phenotypic drug discovery. Sci. Rep. 2019, 9, 5013. [Google Scholar] [CrossRef]
- Vamathevan, J.; Clark, D.; Czodrowski, P.; Dunham, I.; Ferran, E.; Lee, G.; Li, B.; Madabhushi, A.; Shah, P.; Spitzer, M.; et al. Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 2019, 18, 463–477. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Saeed, M.; Kadioglu, O.; Khalid, H.; Sugimoto, Y.; Efferth, T. Activity of the dietary flavonoid, apigenin, against multidrug-resistant tumor cells as determined by pharmacogenomics and molecular docking. J. Nutr. Biochem. 2015, 26, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Seo, E.J.; Kuete, V.; Kadioglu, O.; Krusche, B.; Schroder, S.; Greten, H.J.; Arend, J.; Lee, I.S.; Efferth, T. Antiangiogenic activity and pharmacogenomics of medicinal plants from traditional korean medicine. Evidence-Based Complement. Altern. Med. 2013, 131306. [Google Scholar] [CrossRef]
- Broccatelli, F. QSAR models for P-glycoprotein transport based on a highly consistent data set. J. Chem. Inf. Model. 2012, 52, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Zeino, M.; Saeed, M.E.M.; Kadioglu, O.; Efferth, T. The ability of molecular docking to unravel the controversy and challenges related to P-glycoprotein-a well-known, yet poorly understood drug transporter. Investig. New Drugs 2014, 32, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. Data Warrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Naveja, J.J.; Medina-Franco, J.L. DataWarrior: An evaluation of the open-source drug discovery tool. Expert Opin. Drug Discov. 2019, 14, 335–341. [Google Scholar] [CrossRef]
- Cai, C.P.; Fang, J.S.; Guo, P.F.; Wang, Q.; Hong, H.X.; Moslehi, J.; Cheng, F.X. In silico pharmacoepidemiologic evaluation of drug-induced cardiovascular complications using combined classifiers. J. Chem. Inf. Model. 2018, 58, 943–956. [Google Scholar] [CrossRef]
- Demsar, J.; Curk, T.; Erjavec, A.; Gorup, C.; Hocevar, T.; Milutinovic, M.; Mozina, M.; Polajnar, M.; Toplak, M.; Staric, A.; et al. Orange: Data mining toolbox in python. J. Mach. Learn. Res. 2013, 14, 2349–2353. [Google Scholar]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [Green Version]
- Lo, Y.C.; Rensi, S.E.; Torng, W.; Altman, R.B. Machine learning in chemoinformatics and drug discovery. Drug Discov. Today 2018, 23, 1538–1546. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, J.J.; Han, D.; Zhu, H. From machine learning to deep learning: Progress in machine intelligence for rational drug discovery. Drug Discov. Today 2017, 22, 1680–1685. [Google Scholar] [CrossRef]
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminformatics 2009, 1, 21. [Google Scholar] [CrossRef]
- Cano, G.; Garcia-Rodriguez, J.; Garcia-Garcia, A.; Perez-Sanchez, H.; Benediktsson, J.A.; Thapa, A.; Barr, A. Automatic selection of molecular descriptors using random forest: Application to drug discovery. Expert Syst. Appl. 2017, 72, 151–159. [Google Scholar] [CrossRef]
- Ohashi, R.; Watanabe, R.; Esaki, T.; Taniguchi, T.; Torimoto-Katori, N.; Watanabe, T.; Ogasawara, Y.; Takahashi, T.; Tsukimoto, M.; Mizuguchi, K. Development of simplified in vitro P-glycoprotein substrate assay and in silico prediction models to evaluate transport potential of P-glycoprotein. Mol. Pharm. 2019, 16, 1851–1863. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lee, M.H.; Weng, C.F.; Leong, M.K. Theoretical prediction of the complex P-glycoprotein substrate efflux based on the novel hierarchical support vector regression scheme. Molecules 2018, 23, 1820. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, Q.G.; Li, Y.X.; Tang, L. A new model of flavonoids affinity towards P-glycoprotein: Genetic algorithm-support vector machine with features selected by a modified particle swarm optimization algorithm. Arch. Pharmacal. Res. 2017, 40, 214–230. [Google Scholar] [CrossRef] [PubMed]
- Ngo, T.D.; Tran, T.D.; Le, M.T.; Thai, K.M. Computational predictive models for P-glycoprotein inhibition of in-house chalcone derivatives and drug-bank compounds. Mol. Divers. 2016, 20, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.D.; Chen, J.H.; He, Y.S.; Zhang, Y.L.; Li, G.Y. A method to predict different mechanisms for blood-brain barrier permeability of CNS activity compounds in Chinese herbs using support vector machine. J. Bioinform. Comput. Biol. 2016, 14, 1650005. [Google Scholar] [CrossRef] [PubMed]
- Leong, M.K.; Chen, H.B.; Shih, Y.H. Prediction of promiscuous P-glycoprotein inhibition using a novel machine learning scheme. PLoS ONE 2012, 7, e33829. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, I.; Malik, D.; Jemnitz, K.; Veres, Z.; Hari, P.; Ni, Z.L.; Loo, T.W.; Clarke, D.M.; Hazai, E.; et al. Predicting P-glycoprotein-mediated drug transport based on support vector machine and three-dimensional crystal structure of P-glycoprotein. PLoS ONE 2011, 6, e25815. [Google Scholar] [CrossRef]
- Xue, Y.; Yap, C.W.; Sun, L.Z.; Cao, Z.W.; Wang, J.F.; Chen, Y.Z. Prediction of P-glycoprotein substrates by a support vector machine approach. J. Chem. Inf. Comput. Sci. 2004, 44, 1497–1505. [Google Scholar] [CrossRef]
- Keogh, J.P.; Kunta, J.R. Development, validation and utility of an in vitro technique for assessment of potential clinical drug-drug interactions involving P-glycoprotein. Eur. J. Pharm. Sci. 2006, 27, 543–554. [Google Scholar] [CrossRef]
- Lee, W.Y.; Cheung, C.C.; Liu, K.W.; Fung, K.P.; Wong, J.; Lai, P.B.; Yeung, J.H. Cytotoxic effects of tanshinones from Salvia miltiorrhiza on doxorubicin-resistant human liver cancer cells. J. Nat. Prod. 2010, 73, 854–859. [Google Scholar] [CrossRef]
- Takeshita, H.; Kusuzaki, K.; Ashihara, T.; Gebhardt, M.C.; Mankin, H.J.; Hirasawa, Y. Actin organization associated with the expression of multidrug resistant phenotype in osteosarcoma cells and the effect of actin depolymerization on drug resistance. Cancer Lett. 1998, 126, 75–81. [Google Scholar] [CrossRef]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; Bastos, M.D.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.; Varga, A.; Cherepnev, G.; Radics, R.; Molnar, J.; Ferreira, M.J.U. Apoptosis induction and modulation of P-glycoprotein mediated multidrug resistance by new macrocyclic lathyrane-type diterpenoids. Bioorganic Med. Chem. 2007, 15, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Landau, H.J.; Morrow, M.; Lockerbie, R.O.; Pitts, T.; Eckhardt, S.G. The farnesyl transferase inhibitor, tipifarnib, is a potent inhibitor of the MDR1 gene product, P-glycoprotein, and demonstrates significant cytotoxic synergism against human leukemia cell lines. Leukemia 2007, 21, 739–746. [Google Scholar] [CrossRef]
- Rubin, E.H.; de Alwis, D.P.; Pouliquen, I.; Green, L.; Marder, P.; Lin, Y.; Musanti, R.; Grospe, S.L.; Smith, S.L.; Toppmeyer, D.L.; et al. A phase I trial of a potent P-glycoprotein inhibitor, zosuquidar.3HCl trihydrochloride (LY335979), administered orally in combination with doxorubicin in patients with advanced malignancies. Clin. Cancer Res. 2002, 8, 3710–3717. [Google Scholar]
- Pauli-Magnus, C.; Murdter, T.; Godel, A.; Mettang, T.; Eichelbaum, M.; Klotz, U.; Fromm, M.F. P-glycoprotein-mediated transport of digitoxin, alpha-methyldigoxin and beta-acetyldigoxin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 363, 337–343. [Google Scholar] [CrossRef]
- Vautier, S.; Lacomblez, L.; Chacun, H.; Picard, V.; Gimenez, F.; Farinotti, R.; Fernandez, C. Interactions between the dopamine agonist, bromocriptine and the efflux protein, P-glycoprotein at the blood-brain barrier in the mouse. Eur. J. Pharm. Sci. 2006, 27, 167–174. [Google Scholar] [CrossRef]
- Zhou, L.J.; Chen, X.P.; Gu, Y.Q.; Liang, J.Y. Transport Characteristics of Candesartan in Human Intestinal Caco-2 Cell Line. Biopharmacy and Drug Disposal 2009, 30, 259–264. [Google Scholar] [CrossRef]
- Koizumi, S.; Konishi, M.; Ichihara, T.; Wada, H.; Matsukawa, H.; Goi, K.; Mizutani, S. Flow Cytometric functional analysis of multidrug-resistance by Fluo-3—A comparison with rhodamine-123. Eur. J. Cancer 1995, 31a, 1682–1688. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Smith, C.D. Differential interactions of cytochalasins with P-glycoprotein. Oncol. Res. 1995, 7, 435–443. [Google Scholar] [PubMed]
- Rebbeor, J.F.; Senior, A.E. Effects of cardiovascular drugs on ATPase activity of P-glycoprotein in plasma membranes and in purified reconstituted form. Biophys. Acta (BBA) Biomembr. 1998, 1369, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, S.; Costales, C.; Lazzaro, S.; Eatemadpour, S.; Kimoto, E.; Varma, M.V. Physiologically-based pharmacokinetic modeling approach to predict rifampin-mediated intestinal P-glycoprotein induction. CPT: Pharmacometrics Syst. Pharmacol. 2019, 8, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Lan, L.B.; Sanglard, D.; Furuya, K.; Schuetz, J.D.; Schuetz, E.G. Interaction of cytochrome P450 3A inhibitors with P-glycoprotein. J. Pharmacol. Exp. Ther. 2002, 303, 323–332. [Google Scholar] [CrossRef]
- Takeshita, H.; Kusuzaki, K.; Tsuji, Y.; Hirata, M.; Hashiguchi, S.; Nakamura, S.I.; Murata, H.; Ashihara, T.; Hirasawa, Y. Avoidance of doxorubicin resistance in osteosarcoma cells using a new quinoline derivative, MS-209. Anticancer. Res. 1998, 18, 739–742. [Google Scholar]
- Luo, F.R.; Paranjpe, P.V.; Guo, A.; Rubin, E.; Sinko, P. Intestinal transport of irinotecan in Caco-2 cells and MDCK II cells overexpressing efflux transporters PGP, cMOAT, and MRP1. Drug Metab. Dispos. 2002, 30, 763–770. [Google Scholar] [CrossRef]
- Salvatore, C.A.; Moore, E.L.; Calamari, A.; Cook, J.J.; Michener, M.S.; O’Malley, S.; Miller, P.J.; Sur, C.; Williams, D.L.; Zeng, Z.Z.; et al. Pharmacological properties of MK-3207, a potent and orally active calcitonin gene-related peptide receptor antagonist. J. Pharmacol. Exp. Ther. 2010, 333, 152–160. [Google Scholar] [CrossRef]
- Nemcova-Furstova, V.; Kopperova, D.; Balusikova, K.; Ehrlichova, M.; Brynychova, V.; Vaclavikova, R.; Daniel, P.; Soucek, P.; Kovar, J. Characterization of acquired paclitaxel resistance of breast cancer cells and involvement of ABC transporters. Toxicol. Appl. Pharmacol. 2016, 310, 215–228. [Google Scholar] [CrossRef]
- Shepard, R.L.; Winter, M.A.; Hsaio, S.C.; Pearce, H.L.; Beck, W.T.; Dantzig, A.H. Effect of modulators on the ATPase activity and vanadate nucleotide trapping of human P-glycoprotein. Biochem. Pharmacol. 1998, 56, 719–727. [Google Scholar] [CrossRef]
- Eadie, L.N.; Hughes, T.P.; White, D.L. ABCB1 Overexpression is a key initiator of resistance to tyrosine kinase inhibitors in CML cell lines. PLoS ONE 2016, 11, e0161470. [Google Scholar] [CrossRef]
- Nemethova, V.; Razga, F. Overexpression of ABCB1 as prediction marker for CML: How close we are to translation into clinics? Leukemia 2017, 31, 266–267. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Learning Set | External Validation Set | ||||
|---|---|---|---|---|---|
| Compound | Category | Compound | Category | Compound | Category |
| Escitalopram | Modulator | Hydroxyzine | Non-modulator | Terfenadine | Modulator |
| Simvastatin acid | Modulator | Oxybutynin | Non-modulator | Prazosin | Modulator |
| Neostigmine | Modulator | Ethosuximide | Non-modulator | Prednisone | Modulator |
| Zolmitriptan | Modulator | Warfarin | Non-modulator | Chloroquine | Modulator |
| Atomoxetine | Modulator | Mexilitene | Non-modulator | Lopinavir | Modulator |
| Methysergide | Modulator | Sulpiride | Non-modulator | Prednisolone | Modulator |
| Famciclovir | Modulator | Thiopental | Non-modulator | Vincristine | Modulator |
| Lovastatin acid | Modulator | Lamotrigine | Non-modulator | Sertraline | Modulator |
| Darifenacin | Modulator | Diphenhydramine | Non-modulator | Loperamide | Modulator |
| Paliperidone | Modulator | Enoxacin | Non-modulator | Etoposide | Modulator |
| Trospium | Modulator | Methylphenidate | Non-modulator | Indinavir | Modulator |
| Aprepitant | Modulator | Itraconazole | Non-modulator | Dipyridamole | Modulator |
| Apomorphine | Modulator | Nortriptyline | Non-modulator | Mitoxantrone | Modulator |
| Cetirizine | Modulator | Galantamine | Non-modulator | Cimetidine | Modulator |
| Cyclosporin A | Modulator | Ramelteon | Non-modulator | Bromocriptine | Modulator |
| Labetalol | Modulator | Rivastigmine | Non-modulator | Reserpine | Modulator |
| Amisulpride | Modulator | Ropivacaine | Non-modulator | Oxprenolol | Non-modulator |
| 5-Hydroxymethyl tolterodine | Modulator | Zonisamide | Non-modulator | Alprazolam | Non-modulator |
| Cabergoline | Modulator | Zolpidem | Non-modulator | Oxcarbazepine | Non-modulator |
| Ximelagatran | Modulator | Sulfasalazine | Non-modulator | Tolterodine | Non-modulator |
| Hoechst 33342 | Modulator | Metoclopramide | Non-modulator | Zaleplon | Non-modulator |
| Rhodamine 123 | Modulator | Nalmefene | Non-modulator | Cyclobenzaprine | Non-modulator |
| Actinomycin D | Modulator | Oxycodone | Non-modulator | Nimodipine | Non-modulator |
| Olanzapine | Modulator | Topiramate | Non-modulator | Riluzole | Non-modulator |
| Ranitidine | Modulator | Hydrocodone | Non-modulator | Tiagabine | Non-modulator |
| Astemizole | Modulator | Rosuvastatin | Non-modulator | Nalbuphine | Non-modulator |
| Verapamil | Modulator | Tropisetron | Non-modulator | Duloxetine | Non-modulator |
| Ziprasidone | Modulator | Varenicline | Non-modulator | Pravastatin acid | Non-modulator |
| Chlorpromazine | Modulator | Clemastine | Non-modulator | Promazine | Non-modulator |
| Clozapine | Modulator | Clonazepam | Non-modulator | Bromazepam | Non-modulator |
| Trimethoprim | Modulator | Ropinirole | Non-modulator | Lorazepam | Non-modulator |
| Paroxetine | Modulator | Solifenacin | Non-modulator | Mirtazapine | Non-modulator |
| Learning Set | External Validation Set | ||||||
|---|---|---|---|---|---|---|---|
| Compound | Category | Compound | Category | Compound | Category | Compound | Category |
| Ginsenoside | Inhibitor | Epirubicin | Substrate | Agosterol | Inhibitor | Colchicin | Substrate |
| Laniquidar | Inhibitor | Etoposide | Substrate | Amiodarone | Inhibitor | Dexamethazone | Substrate |
| Loratidine | Inhibitor | Fexofenadine | Substrate | Amorinin | Inhibitor | Digoxin | Substrate |
| Mibefradil | Inhibitor | Hoechst 33342 | Substrate | Apigenin | Inhibitor | Docetaxel | Substrate |
| Naringenin | Inhibitor | Idarubicin | Substrate | Atorvastatin | Inhibitor | Doxorubicin | Substrate |
| Pgp-4008 | Inhibitor | Irinotecan | Substrate | Atovaquone | Inhibitor | Daunorubicin | Substrate |
| Phloretin | Inhibitor | Kaempferol | Substrate | Biochanin | Inhibitor | ||
| Quercetin | Inhibitor | Loperamide | Substrate | Biricodar | Inhibitor | ||
| Quinine | Inhibitor | Mitomycin | Substrate | Catechin | Inhibitor | ||
| Rotenone | Inhibitor | Mitoxantrone | Substrate | Cefoperazone | Inhibitor | ||
| Sakuranetin | Inhibitor | Ondansetron | Substrate | Chrysine | Inhibitor | ||
| Sertraline | Inhibitor | Paclitaxel | Substrate | Cyclosporine | Inhibitor | ||
| Sinensetin | Inhibitor | Procyanidin B2 | Substrate | Diltiazem | Inhibitor | ||
| Stigmasterol | Inhibitor | Rhodamine 123 | Substrate | Elacridar | Inhibitor | ||
| Syringaresinol | Inhibitor | Tenoposide | Substrate | ||||
| Tamoxifen | Inhibitor | Topotecan | Substrate | ||||
| Tariquidar | Inhibitor | Vinblastine | Substrate | ||||
| Valspodar | Inhibitor | Vincristine | Substrate | ||||
| Verapamil | Inhibitor | Vindesine | Substrate | ||||
| Zosuquidar | Inhibitor | Vinorelbine | Substrate | ||||
| Steps | Sensitivity | Specificity | Overall Predictive Accuracy | Precision |
|---|---|---|---|---|
| Learning | 0.938 | 0.969 | 0.953 | 0.968 |
| External Validation | 0.938 | 0.938 | 0.938 | 0.938 |
| Steps | Sensitivity | Specificity | Overall Predictive Accuracy | Precision |
|---|---|---|---|---|
| Learning | 0.750 | 0.700 | 0.725 | 0.714 |
| External Validation | 0.786 | 0.833 | 0.800 | 0.917 |
| x | y | z | |
|---|---|---|---|
| Number of Points | 126 | 98 | 116 |
| Grid Center | 168.614 | 166.372 | 162.000 |
| Grid Spacing (Å) | 0.375 |
| Name | ChEMBL ID | Inhibitor Probability | Class | VINA LBE (kcal/mol) |
|---|---|---|---|---|
| Karavoate P | CHEMBL1641677 | 0.849 | Synthetic | −12.200 ± 1.212 |
| Tribenzoylbalsaminol F | CHEMBL1928854 | 0.549 | Synthetic | −12.033 ± 0.896 |
| Zosuquidar | CHEMBL444172 | 0.513 | Synthetic | −11.967 ± 0.058 |
| Latilagascenes D | CHEMBL435917 | 0.566 | Synthetic | −11.700 ± 0.001 |
| Dihydrocytochalasin B | CHEMBL2074735 | 0.513 | Synthetic | −11.367 ± 0.231 |
| Jolkinoate I | CHEMBL2315618 | 0.593 | Synthetic | −11.300 ± <0.001 |
| Karavoate K | CHEMBL1641672 | 0.849 | Synthetic | −11.267 ± 0.493 |
| Fanchinin | CHEMBL176045 | 0.586 | Synthetic | −11.233 ± 0.208 |
| Latilagascene I | CHEMBL511018 | 0.586 | Synthetic | −11.167 ± 0.058 |
| Karavoate L | CHEMBL1641673 | 0.766 | Synthetic | −11.133 ± 0.808 |
| 3-Methylcholanthrene | CHEMBL40583 | 0.788 | Synthetic | −11.100 ± <0.001 |
| Lonafarnib | CHEMBL298734 | 0.567 | Synthetic | −11.000 ± <0.001 |
| Karavoate N | CHEMBL1641675 | 0.666 | Synthetic | −10.933 ± 0.058 |
| Tariquidar | CHEMBL348475 | 0.619 | Synthetic | −10.933 ± 0.404 |
| Pimozide | CHEMBL1423 | 0.517 | Synthetic | −10.900 ± 0.100 |
| Karavoate I | CHEMBL1641670 | 0.766 | Synthetic | −10.767 ± 0.058 |
| Cryptotanshinone | CHEMBL187460 | 0.663 | Natural | −10.700 ± <0.001 |
| Jolkinol B | CHEMBL489265 | 0.577 | Synthetic | −10.700 ± <0.001 |
| Astemizole | CHEMBL296419 | 0.617 | Synthetic | −10.667 ± 0.115 |
| Metergoline | CHEMBL19215 | 0.732 | Natural | −10.600 ± <0.001 |
| Name | ChEMBL ID | Substrate probability | Class | VINA LBE (kcal/mol) |
|---|---|---|---|---|
| Vindoline | CHEMBL526546 | 0.771 | Synthetic | −15.000 ± <0.001 |
| Cepharanthin | CHEMBL2074948 | 0.614 | Natural | −12.600 ± <0.001 |
| Latilagascene G | CHEMBL448193 | 0.514 | Synthetic | −12.300 ± <0.001 |
| Mk3207 | CHEMBL1910936 | 0.733 | Synthetic | −12.167 ± 0.058 |
| Ergocristine | CHEMBL446315 | 0.767 | Natural | −12.067 ± 0.058 |
| Cytochalasin E | CHEMBL494856 | 0.6 | Natural | −11.800 ± <0.001 |
| Jolkinoate L | CHEMBL2315621 | 0.567 | Synthetic | −11.533 ± 0.058 |
| Irinotecan | CHEMBL481 | 0.967 | Natural | −11.400 ± 0.819 |
| Latilagascenes E | CHEMBL373511 | 0.614 | Synthetic | −11.367 ± 0.116 |
| Dofequidar | CHEMBL65067 | 0.583 | Synthetic | −11.300 ± 0.001 |
| Acetyldigoxin | CHEMBL2074725 | 0.708 | Natural | −11.233 ± 0.808 |
| Dihydroergocristine | CHEMBL601773 | 0.767 | Natural | −11.133 ± 0.666 |
| Telcagepant | CHEMBL236593 | 0.517 | Synthetic | −11.067 ± 0.058 |
| Ergotamine | CHEMBL442 | 0.8 | Natural | −10.933 ± 0.058 |
| Candesartan Cilexetil | CHEMBL1014 | 0.567 | Synthetic | −10.900 ± 0.200 |
| Digoxin | CHEMBL1751 | 0.708 | Natural | −10.833 ± 1.097 |
| Bromocriptine | CHEMBL493 | 0.767 | Natural | −10.800 ± 0.100 |
| Itrazole | CHEMBL64391 | 0.564 | Synthetic | −10.700 ± 0.436 |
| Digitoxin | CHEMBL254219 | 0.725 | Natural | −10.667 ± 0.462 |
| Paclitaxel | CHEMBL428647 | 0.808 | Natural | −10.633 ± 0.462 |
| P-gp Inhibitor | AutoDock LBE (kcal/mol) | Predicted Inhibition Constant (µM) |
|---|---|---|
| 3-Methylcholanthrene | −8.900 ± 0.001 | 0.300 ± <0.001 |
| Astemizole | −9.693 ± 0.047 | 0.079 ± 0.007 |
| Cryptotanshinone | −9.010 ± 0.001 | 0.251 ± <0.001 |
| Dihydrocytochalasin B | −10.460 ± 0.020 | 0.0212 ± 0.001 |
| Fanchinin | −9.937 ± 0.067 | 0.0522 ± 0.006 |
| Jolkinoate I | −10.440 ± 0.200 | 0.0232 ± 0.008 |
| Jolkinol B | −10.250 ± 0.044 | 0.0307 ± 0.002 |
| Karavoate I | −12.310 ± 0.235 | 0.001 ± <0.001 |
| Karavoate K | −12.330 ± 0.213 | 0.001 ± <0.001 |
| Karavoate L | −12.807 ± 0.200 | 0.0004 ± <0.001 |
| Karavoate N | −12.160 ± 0.560 | 0.002 ± 0.001 |
| Karavoate P | −13.537 ± 0.605 | 0.0002 ± <0.001 |
| Latilagascene I | −11.147 ± 0.561 | 0.009 ± 0.009 |
| Latilagascenes D | −12.220 ± 0.370 | 0.001 ± 0.001 |
| Lonafarnib | −11.433 ± 0.087 | 0.004 ± 0.001 |
| Metergoline | −9.737 ± 0.029 | 0.073 ± 0.004 |
| Pimozide | −10.220 ± 0.324 | 0.031 ± 0.025 |
| Tariquidar | −11.273 ± 0.274 | 0.006 ± 0.002 |
| Tribenzoylbalsaminol F | −12.403 ± 0.118 | 0.001 ± <0.001 |
| Zosuquidar | −11.257 ± 0.361 | 0.006 ± 0.004 |
| Elacridar (positive control) | −11.093 ± 0.361 | 0.008 ± 0.004 |
| P-gp substrate | AutoDock LBE (kcal/mol) | Predicted Inhibition Constant (µM) |
|---|---|---|
| Acetyldigoxin | −11.767 ± 0.480 | 0.003 ± 0.002 |
| Bromocriptine | −12.360 ± 1.02 | 0.002 ± 0.001 |
| Candesartan Cilexetil | −11.153 ± 0.370 | 0.007 ± 0.004 |
| Cepharanthin | −10.753 ± 0.006 | 0.013 ± <0.001 |
| Cytochalasin E | −10.957 ± 0.006 | 0.093 ± 0.001 |
| Digitoxin | −11.390 ± 0.517 | 0.006 ± 0.004 |
| Digoxin | −11.500 ± 0.151 | 0.004 ± 0.001 |
| Dihydroergocristine | −11.670 ± 0.056 | 0.003 ± <0.001 |
| Dofequidar | −10.970 ± 0.351 | 0.010 ± 0.006 |
| Ergocristine | −12.407 ± 0.012 | 0.001 ± <0.001 |
| Ergotamine | −11.227 ± 0.150 | 0.006 ± 0.001 |
| Irinotecan | −11.380 ± 0.020 | 0.005 ± <0.001 |
| Itrazole | −10.843 ± 0.186 | 0.012 ± 0.003 |
| Jolkinoate L | −10.643 ± 0.681 | 0.022 ± 0.016 |
| Latilagascenes E | −11.770 ± 0.185 | 0.002 ± 0.001 |
| Latilagescene G | −12.500 ± 0.316 | 0.001 ± <0.001 |
| Mk-3207 | −11.650 ± 0.020 | 0.003 ± <0.001 |
| Paclitaxel | −9.607 ± 0.359 | 0.103 ± 0.065 |
| Telcagepant | −9.333 ± 0.021 | 0.144 ± 0.005 |
| Vindoline | −7.337 ± 0.211 | 4.363 ± 1.389 |
| Doxorubicin (positive control) | −11.070 ± 0.135 | 0.008 ± 0.002 |
| P-gp Inhibitor | AutoDock LBE (kcal/mol) | Predicted Inhibition Constant (µM) |
|---|---|---|
| Oxprenolol | −5.743 ± 0.398 | 70.273 ± 40.057 |
| Promazine | −6.933 ± 0.021 | 8.273 ± 0.262 |
| Riluzole | −5.380 ± 0.010 | 114.080 ± 2.326 |
| Descriptor | Inhibitor | Substrate |
|---|---|---|
| cLogP | 3.498 ± 2.464 | 3.134 ± 2.962 |
| Total surface area | 311.199 ± 188.142 | 461.870 ± 286.187 |
| Shape index | 0.529 ± 0.125 | 0.429 ± 0.081 |
| Molecular flexibility | 0.395 ± 0.141 | 0.332 ± 0.114 |
| Rotatable bonds | 6.799 ± 12.158 | 9.818 ± 11.778 |
| Aromatic rings | 1.450 ± 1.168 | 1.918 ± 1.330 |
| Aromatic atoms | 8.237 ± 6.470 | 10.759 ± 7.098 |
| Symmetric atoms | 2.649 ± 3.637 | 3.582 ± 4.477 |
| Aromatic nitrogens | 0.301 ± 0.772 | 0.559 ± 1.141 |
| Basic nitrogens | 0.441 ± 0.625 | 0.659 ± 0.762 |
| Acidic oxygens | 0.117 ± 0.361 | 0.171 ± 0.462 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kadioglu, O.; Efferth, T. A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells 2019, 8, 1286. https://doi.org/10.3390/cells8101286
Kadioglu O, Efferth T. A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells. 2019; 8(10):1286. https://doi.org/10.3390/cells8101286
Chicago/Turabian StyleKadioglu, Onat, and Thomas Efferth. 2019. "A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking" Cells 8, no. 10: 1286. https://doi.org/10.3390/cells8101286
APA StyleKadioglu, O., & Efferth, T. (2019). A Machine Learning-Based Prediction Platform for P-Glycoprotein Modulators and Its Validation by Molecular Docking. Cells, 8(10), 1286. https://doi.org/10.3390/cells8101286