Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients

 ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Considerations
2.2. Patients
2.3. Bacterial Strains
2.4. MDM Isolation and Culture
2.5. MDM Internalization and Survival Assays
2.6. Enzyme-Linked Immunosorbent Assay
2.7. Short-Term Silencing of ULK-1 and IRGM Genes
2.8. Western Blotting
2.9. Sample Size Calculation
2.10. Data Collection and Statistical Analysis
3. Results
3.1. Clinical and Genetics Characteristics of the Cohort
3.2. The Internalization of AIEC Bacteria Was Not Influenced by the Origin of the Monocytes-Derived Macrophages but Was Lower in Patients Carrying the CD-Associated Polymorphism CYLD
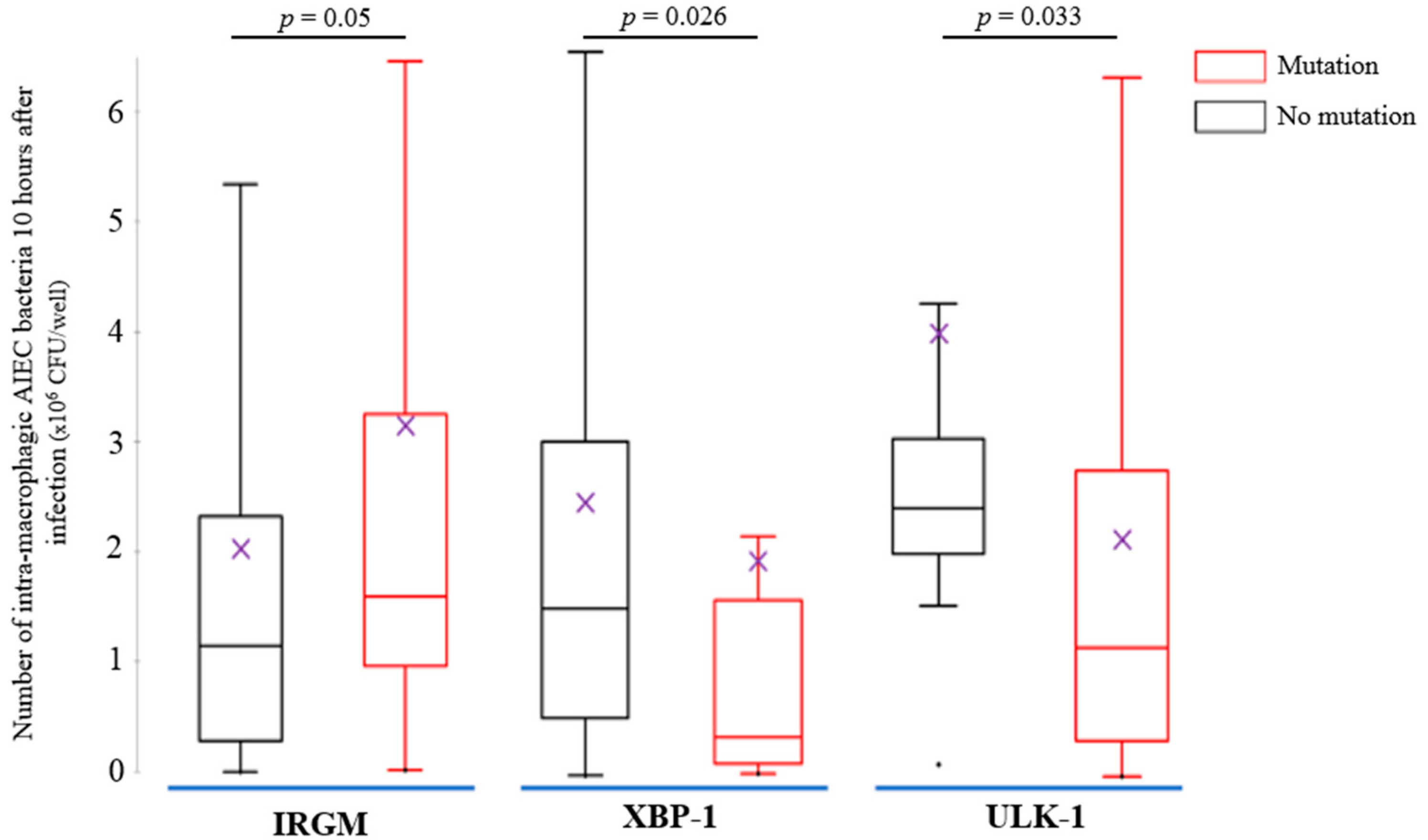
3.3. AIEC Bacteria Survival Is Increased within MDM from CD Patients and Was Impacted by the CD-Associated Polymorphisms IRGM, XBP-1 and ULK-1
3.4. AIEC Bacteria Induce an Excessive and Disordered Inflammatory Response by Macrophages
3.5. ULK-1 Protein Activity and AIEC Bacteria Survival Are Dependent of Each Other in MDM from CD Patients
3.6. The Internalization of AIEC Bacteria is Influenced by the Autophagic Process and Especially the Expression of ULK-1 in MDM
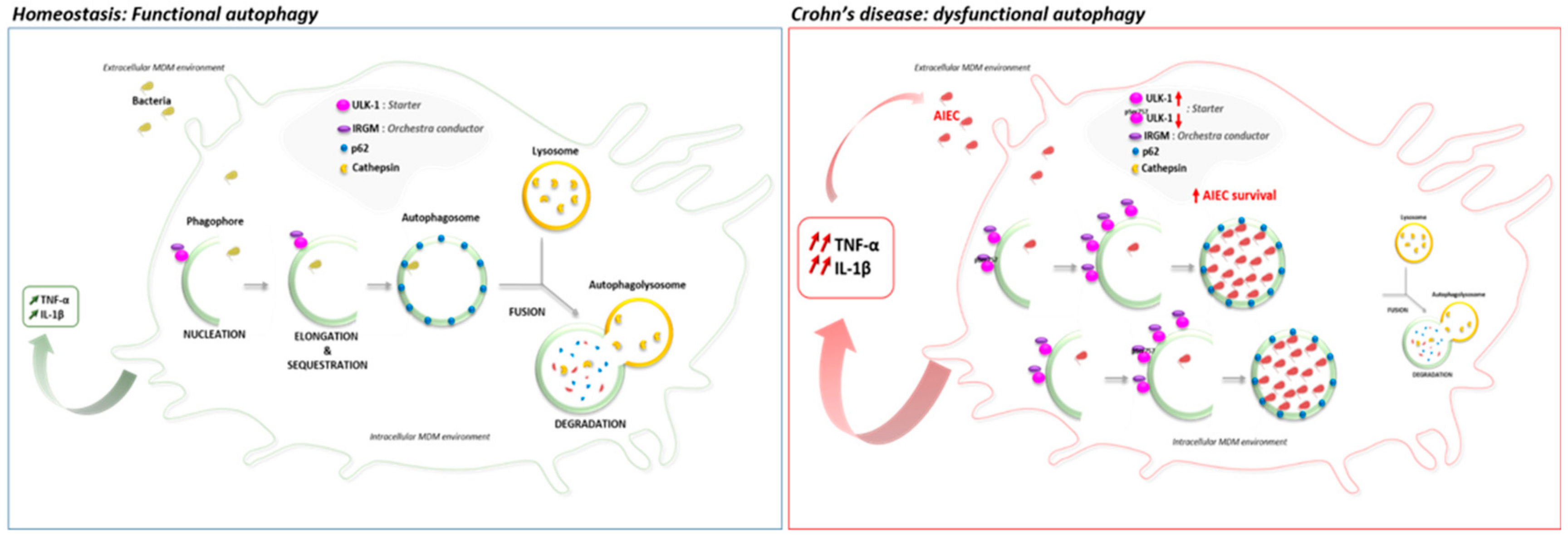
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Peyrin-Biroulet, L.; Loftus, E.V.; Colombel, J.-F.; Sandborn, W.J. The natural history of adult Crohn’s disease in population-based cohorts. Am. J. Gastroenterol. 2010, 105, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Cieza, A.; Sandborn, W.J.; Coenen, M.; Chowers, Y.; Hibi, T.; Kostanjsek, N.; Stucki, G.; Colombel, J. The International Programme to Develop New Indexes for Crohn’s Disease (IPNIC) Group. Development of the first disability index for inflammatory bowel disease based on the international classification of functioning, disability and health. Gut 2012, 61, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Seksik, P.; Rigottier-Gois, L.; Gramet, G.; Sutren, M.; Pochart, P.; Marteau, P.; Jian, R.; Doré, J. Alterations of the dominant faecal bacterial groups in patients with Crohn’s disease of the colon. Gut 2003, 52, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Massier, S.; Darfeuille-Michaud, A.; Billard, E.; Barnich, N. Understanding host-adherent-invasive Escherichia coli interaction in Crohn’s disease: Opening up new therapeutic strategies. BioMed Res. Int. 2014, 2014, 567929. [Google Scholar] [CrossRef] [PubMed]
- Darfeuille-Michaud, A.; Neut, C.; Barnich, N.; Lederman, E.; Di Martino, P.; Desreumaux, P.; Gambiez, L.; Joly, B.; Cortot, A.; Colombel, J. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology 1998, 115, 1405–1413. [Google Scholar] [CrossRef]
- Darfeuille-Michaud, A.; Boudeau, J.; Bulois, P.; Neut, C.; Glasser, A.L.; Barnich, N.; Bringer, M.; Swidsinski, A.; Beaugerie, L.; Colombel, J. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 2004, 127, 412–421. [Google Scholar] [CrossRef]
- Bringer, M.-A.; Glasser, A.-L.; Tung, C.-H.; Méresse, S.; Darfeuille-Michaud, A. The Crohn’s disease-associated adherent-invasive Escherichia coli strain LF82 replicates in mature phagolysosomes within J774 macrophages. Cell. Microbiol. 2006, 8, 471–484. [Google Scholar] [CrossRef]
- Bringer, M.-A.; Billard, E.; Glasser, A.-L.; Colombel, J.-F.; Darfeuille-Michaud, A. Replication of Crohn’s disease-associated AIEC within macrophages is dependent on TNF-α secretion. Lab. Investig. 2012, 92, 411–419. [Google Scholar] [CrossRef]
- De la Fuente, M.; Franchi, L.; Araya, D.; Díaz-Jiménez, D.; Olivares, M.; Álvarez-Lobos, M.; Golenbock, D.; González, M.; López-Kostner, F.; Quera, R.; et al. Escherichia coli isolates from inflammatory bowel diseases patients survive in macrophages and activate NLRP3 inflammasome. Int. J. Med. Microbiol. IJMM 2014, 304, 384–392. [Google Scholar] [CrossRef]
- Dunne, K.A.; Allam, A.; McIntosh, A.; Houston, S.A.; Cerovic, V.; Goodyear, C.S.; Roe, A.J.; Beatson, S.A.; Milling, S.W.; Walker, D.; et al. Increased S-Nitrosylation and Proteasomal Degradation of Caspase-3 during Infection Contribute to the Persistence of Adherent Invasive Escherichia coli (AIEC) in Immune Cells. PLoS ONE 2013, 8, e68386. [Google Scholar] [CrossRef]
- Glasser, A.L.; Boudeau, J.; Barnich, N.; Perruchot, M.H.; Colombel, J.F.; Darfeuille-Michaud, A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001, 69, 5529–5537. [Google Scholar] [CrossRef] [PubMed]
- Lapaquette, P.; Bringer, M.-A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell. Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Roberts, C.L.; Hart, C.A.; Martin, H.M.; Edwards, S.W.; Rhodes, J.M.; Campbell, B.J. Replication of Colonic Crohn’s Disease Mucosal Escherichia coli Isolates within Macrophages and Their Susceptibility to Antibiotics. Antimicrob. Agents Chemother. 2008, 52, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Vazeille, E.; Buisson, A.; Bringer, M.A.; Goutte, M.; Ouchchane, L.; Hugot, J.P.; De Vallée, A.; Barnich, N.; Bommelaer, G.; Darfeuille-Michaud, A. Monocyte-derived macrophages from Crohn’s disease patients are impaired in the ability to control intracellular adherent-invasive Escherichia coli and exhibit disordered cytokine secretion profile. J. Crohns. Colitis 2015, 9, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.R.; Hudspith, B.N.; Rayment, N.B.; Prescott, N.J.; Petrovska, L.; Hermon-Taylor, J.; Brostoff, J.; Boussioutas, A.; Mathew, C.G.; Sanderson, J.D. Defective macrophage handling of Escherichia coli in Crohn’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1265–1274. [Google Scholar] [CrossRef]
- Cho, J.H.; Brant, S.R. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology 2011, 140, 1704–1712. [Google Scholar] [CrossRef]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.; Hébuterne, X. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Sadabad, M.S.; Regeling, A.; De Goffau, M.C.; Blokzijl, T.; Weersma, R.K.; Penders, J.; Faber, K.N.; Harmsen, H.J.M.; Dijkstra, G. The ATG16L1-T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut 2015, 64, 1546–1552. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; Van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1392–1397. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleynen, I.; Vazeille, E.; Artieda, M.; Verspaget, H.W.; Szczypiorska, M.; Bringer, M.A. Genetic and microbial factors modulating the ubiquitin proteasome system in in fl ammatory bowel disease. Gut 2014, 63, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De la Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef] [Green Version]
- Manzoni, C. LRRK2 and autophagy: A common pathway for disease. Biochem. Soc. Trans. 2012, 40, 1147–1151. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Edward, E.S.; Nieuwenhuis, D.E.H.; Higgins, D.E.; Schreiber, S.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk of human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Cleynen, I.; González, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlík, M.; Crusius, B.J.A.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [PubMed] [Green Version]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Marks, D.J.; Harbord, M.W.; MacAllister, R.; Rahman, F.Z.; Young, J.; Al-Lazikani, B.; Lees, W.; Path, M.N.M.R.C.; Bloom, S.; Segal, A.W. Defective acute inflammation in Crohn’s disease: A clinical investigation. Lancet 2006, 367, 668–678. [Google Scholar] [CrossRef]
- Smith, A.M.; Rahman, F.Z.; Hayee, B.H.; Graham, S.J.; Marks, D.J.; Sewell, G.W.; Palmer, C.D.; Wilde, J.; Foxwell, B.M.J.; Gloger, I.S.; et al. Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J. Exp. Med. 2009, 206, 1883–1897. [Google Scholar] [CrossRef] [PubMed]
- Campos, N.; Magro, F.; Castro, A.R.; Cabral, J.; Rodrigues, P.; Silva, R.; Appelberg, R.; Rodrigues, S.; Lopes, S.; Macedo, G.; et al. Macrophages from IBD patients exhibit defective tumour necrosis factor-α secretion but otherwise normal or augmented pro-inflammatory responses to infection. Immunobiology 2011, 216, 961–970. [Google Scholar] [CrossRef]
- Sewell, G.W.; Rahman, F.Z.; Levine, A.P.; Jostins, L.; Smith, P.J.; Walker, A.P.; Bloom, S.L.; Segal, A.W.; Smith, A.M. Defective tumor necrosis factor release from Crohn’s disease macrophages in response to toll-like receptor activation: Relationship to phenotype and genome-wide association susceptibility loci. Inflamm. Bowel Dis. 2012, 18, 2120–2127. [Google Scholar] [CrossRef]
- Smith, A.M.; Sewell, G.W.; Levine, A.P.; Chew, T.S.; Dunne, J.; O’Shea, N.R.; Smith, P.J.; Harrison, P.J.; Macdonald, C.M.; Bloom, S.L. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology 2014, 144, 45–55. [Google Scholar] [CrossRef]
- Flanagan, P.K.; Chiewchengchol, D.; Wright, H.L.; Edwards, S.W.; Alswied, A.; Satsangi, J.; Subramanian, S.; Rhodes, J.M.; Campbell, B.J. Killing of Escherichia coli by Crohn’s Disease Monocyte-derived Macrophages and Its Enhancement by Hydroxychloroquine and Vitamin D. Inflamm. Bowel Dis. 2015, 21, 1499–1510. [Google Scholar] [CrossRef]
- Peyrin-Biroulet, L.; Reinisch, W.; Colombel, J.F.; Mantzaris, G.J.; Kornbluth, A.; Diamond, R.; Rutgeerts, P.; Tang, L.K.; Cornillie, F.J.; Sandborn, W.J. Clinical disease activity, C-reactive protein normalisation and mucosal healing in Crohn’s disease in the SONIC trial. Gut 2014, 63, 88–95. [Google Scholar] [CrossRef]
- Schwarzmaier, D.; Foell, D.; Weinhage, T.; Varga, G.; Däbritz, J. Peripheral monocyte functions and activation in patients with quiescent Crohn’s disease. PLoS ONE 2013, 8, e62761. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, F.; Monteleone, I.; Sarra, M.; Calabrese, E.; Marafini, I.; Cretella, M.; Sedda, S.; Biancone, L.; Pallone, F.; Monteleone, G. Distinct Profiles of Effector Cytokines Mark the Different Phases of Crohn’s Disease. PLoS ONE 2013, 8, e54562. [Google Scholar] [CrossRef] [PubMed]
- Caprilli, R.; Frieri, G. The dyspeptic macrophage 30 years later: An update in the pathogenesis of Crohn’s disease. Dig. Liver Dis. Off. J. Ital. Soc. Gastroenterol. Ital. Assoc. Study Liver 2009, 41, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Okabe, N.; Ikura, S.; Uchida, Y.; Fujita, K.; Yao, T. Immunological studies on Crohn’s disease. VIII. Diminished phagocytic activity in peripheral blood monocytes. J. Clin. Lab. Immunol. 1990, 32, 29–31. [Google Scholar] [PubMed]
- Denizot, J.; Sivignon, A.; Barreau, F.; Darcha, C.; Chan, C.H.; Stanners, C.P.; Hofman, P.; Darfeuille-Michaud, A.; Barnich, N. Adherent-invasive Escherichia coli Induce Claudin-2 Expression and Barrier Defect in CEABAC10 Mice and Crohn’ s Disease Patients. Inflamm. Bowel Dis. 2012, 18, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterology 2006, 130, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Low, D.; Tran, H.T.; Lee, I.A.; Dreux, N.; Kamba, A.; Reinecker, H.C.; Darfeuille-Michaud, A.; Barnich, N.; Mizoguchi, E. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 2013, 145, 602–612. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD Patients | UC Patients | Healthy Subjects | |
|---|---|---|---|
| Age, mean (±SD) | 41.3 ± 13.9 | 43.1 ± 14.6 | 33.6 ± 10.8 |
| Disease duration at inclusion, mean (±SD) | 11.1 ± 9.8 | 9.9 ± 9.1 | - |
| Body Mass Index, mean (±SD) | 24.6 ± 5.5 | 24.4 ± 4.6 | 22.5 ± 2.5 |
| Male gender, n (%) | 32 (33.7) | 17 (56.7) | 5 (33.3) |
| Tobacco use, n (%) | |||
| Non-smokers, n (%) | 61 (74.2) | 9 (30.0) | 11 (73.3) |
| Former smokers, n (%) | 6 (6.3) | 18 (60.0) | 1 (6.7) |
| Active smokers, n (%) | 28 (29.5) | 3 (10.0) | 3 (20.0) |
| Previous intestinal resection, n (%) | 30 (31.6) | 0 (0.0) | 0 (0.0) |
| Previous appendectomy, n (%) | 35 (36.8) | 4 (13.3) | 2 (13.3) |
| Familial history of IBD, n (%) | 12 (12.6) | 4 (13.3) | 0 (0.0) |
| Montreal Classification | |||
| Age at diagnosis | |||
| A1, n (%) | 5 (5.3) | - | - |
| A2, n (%) | 71 (74.7) | - | - |
| A3, n (%) | 19 (20.0) | - | - |
| Disease location | - | ||
| L1, n (%) | 26 (27.4) | - | - |
| L2, n (%) | 21 (22.1) | - | - |
| L3, n (%) | 48 (50.5) | - | - |
| L4, n (%) | 4 (4.2) | - | - |
| Behaviour | - | ||
| B1, n (%) | 42 (44.2) | - | - |
| B2, n (%) | 22 (23.2) | - | - |
| B3, n (%) | 29 (30.5) | - | - |
| Extension | |||
| E1, n (%) | - | 3 (10.0) | - |
| E2, n (%) | - | 14 (46.7) | - |
| E3, n (%) | - | 13 (43.3) | - |
| History of perianal lesions, n (%) | 40 (42.1) | - | - |
| Concomittant therapies | |||
| 5-ASA, n (%) | 2 (2.1) | 7 (23.3) | - |
| Corticosteroids, n (%) | 6 (6.3) | 2 (6.6) | - |
| Budesonide, n (%) | 4 (4.2) | 0 (0.0) | - |
| Thiopurines, n (%) | 32 (33.7) | 8 (26.7) | - |
| Methotrexate, n (%) | 10 (10.5) | 0 (0.0) | - |
| Infliximab, n (%) | 40 (42.1) | 16 (53.3) | - |
| Adalimumab, n (%) | 16 (16.3) | 2 (6.7) | - |
| Golimumab, n (%) | 0 (0.0) | 1 (3.3) | - |
| Ustekinumab, n (%) | 1 (1.1) | 0 (0.0) | - |
| Vedolizumab, n (%) | 8 (8.4) | 4 (13.3) | - |
| CDAI, median (IQR) | 80.0 (24.0–184.0) | - | - |
| SCCAI, median (IQR) | - | 2 (0.5–4.8) | - |
| Partial Mayo score, median (IQR) | - | 1 (0–4) | - |
| Leucocytes, (Giga/L), median (IQR) | 7.28 (5.69–9.55) | 6.13 (5.12–7.89) | 7.34 (6.30–8.72) |
| Neutrophils, (Giga/L), median (IQR) | 4.41 (3.21–5.75) | 3.21 (2.49-4.34) | 4.13 (3.49–5.59) |
| Monocytes, (Giga/L), median (IQR) | 0.57 (0.43–0.77) | 0.53 (0.42–0.62] | 0.50 (0.47–0.68) |
| Platelets, (cells ×103/L), median (IQR) | 292 (242–369) | 283 (242–378) | 250 (222–270) |
| CRP, (mg/L), median (IQR) | 3.5 (2.9–12.0) | 3.0 (1.7–8.1) | 2.9 (2.9–2.9) |
| Faecal Calprotectin, (µg/g) median (IQR) | 328 (100–1380) | 433 (100–780] | - |
| Vitamin-D, (µg/L) median (IQR) | 20.1 (13.4–27.5) | - | - |
| Crohn’s Disease Patients n = 95 | Ulcerative Colitis Patients n = 30 | Healthy Controls n = 15 | p-Value | |
|---|---|---|---|---|
| NOD2 | ||||
| rs2066844 (snp8) | 21 (22.1%) | 2 (6.7%) | 2 (13.3%) | 0.14 |
| rs2066845 (snp12) | 8 (8.4%) | 3 (10.0%) | 0 (0.0%) | 0.47 |
| rs2066847 (snp13) | 16 (16.8%) | 1 (3.3%) | 0 (0.0%) | 0.044 * |
| ATG16L1 | 77 (81.1%) | 25 (83.3%) | 9 (60.0%) | 0.14 |
| IRGM | 27 (28.4%) | 5 (16.7%) | 5 (33.3%) | 0.36 |
| ULK1 | 82 (86.3%) | 27 (90.0%) | 13 (86.6%) | 0.87 |
| LRRK2 | 7 (7.3%) | 2 (6.7%) | 1 (6.7%) | 0.98 |
| XBP1 | 13 (13.7%) | 5 (16.7%) | 1 (6.7%) | 0.65 |
| CYLD | ||||
| rs17314544 | 56 (58.9%) | 21 (70.0%) | 10 (66.7%) | 0.51 |
| rs2302759 | 93 (97.8%) | 29 (96.7%) | 14 (93.3%) | 0.60 |
| rs7205423 | 66 (69.5%) | 22 (73.3%) | 12 (80.0%) | 0.68 |
| rs12324931 | 21 (22.1%) | 2 (6.7%) | 2 (13.3%) | 0.14 |
| USP40 | ||||
| rs12472244 | 35 (36.8%) | 8 (26.7%) | 6 (40.0%) | 0.54 |
| rs4047198 | 88 (92.6%) | 28 (93.3%) | 15 (100.0%) | 0.55 |
| rs838548 | 88 (92.6%) | 28 (93.3%) | 15 (100.0%) | 0.55 |
| MDM from CD Patients | Spearman R | p-Value | |
|---|---|---|---|
| IL-1β | LF82 | 0.5398 | <0.0001 |
| K-12 | 0.08662 | 0.4449 | |
| IL-6 | LF82 | 0.287 | 0.0061 |
| K-12 | −0.04959 | 0.6622 | |
| IL-10 | LF82 | 0.4638 | <0.0001 |
| K-12 | 0.02859 | 0.8025 | |
| TNF-α | LF82 | 0.3142 | 0.0026 |
| K-12 | 0.01768 | 0.8763 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buisson, A.; Douadi, C.; Ouchchane, L.; Goutte, M.; Hugot, J.-P.; Dubois, A.; Minet-Quinard, R.; Bouvier, D.; Bommelaer, G.; Vazeille, E.; et al. Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients. Cells 2019, 8, 1394. https://doi.org/10.3390/cells8111394
Buisson A, Douadi C, Ouchchane L, Goutte M, Hugot J-P, Dubois A, Minet-Quinard R, Bouvier D, Bommelaer G, Vazeille E, et al. Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients. Cells. 2019; 8(11):1394. https://doi.org/10.3390/cells8111394
Chicago/Turabian StyleBuisson, Anthony, Clara Douadi, Lemlih Ouchchane, Marion Goutte, Jean-Pierre Hugot, Anaëlle Dubois, Régine Minet-Quinard, Damien Bouvier, Gilles Bommelaer, Emilie Vazeille, and et al. 2019. "Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients" Cells 8, no. 11: 1394. https://doi.org/10.3390/cells8111394
APA StyleBuisson, A., Douadi, C., Ouchchane, L., Goutte, M., Hugot, J. -P., Dubois, A., Minet-Quinard, R., Bouvier, D., Bommelaer, G., Vazeille, E., & Barnich, N. (2019). Macrophages Inability to Mediate Adherent-Invasive E. coli Replication is Linked to Autophagy in Crohn’s Disease Patients. Cells, 8(11), 1394. https://doi.org/10.3390/cells8111394