The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis


Abstract
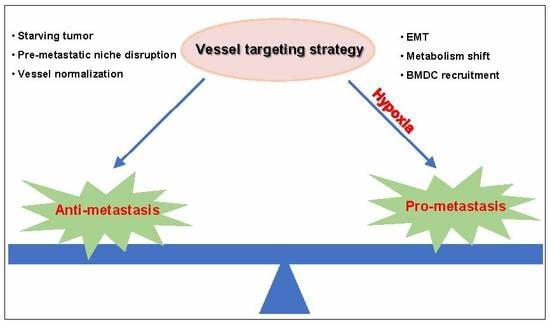
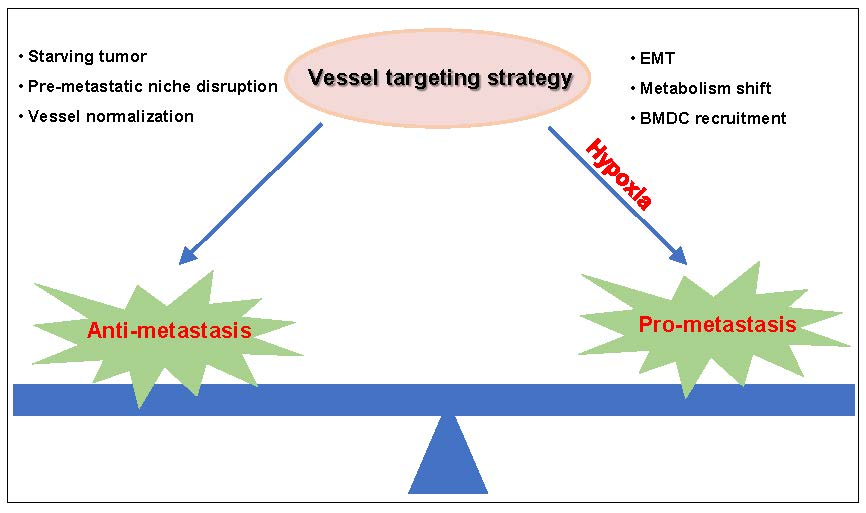
:
1. Introduction
2. Angiogenesis and Metastasis
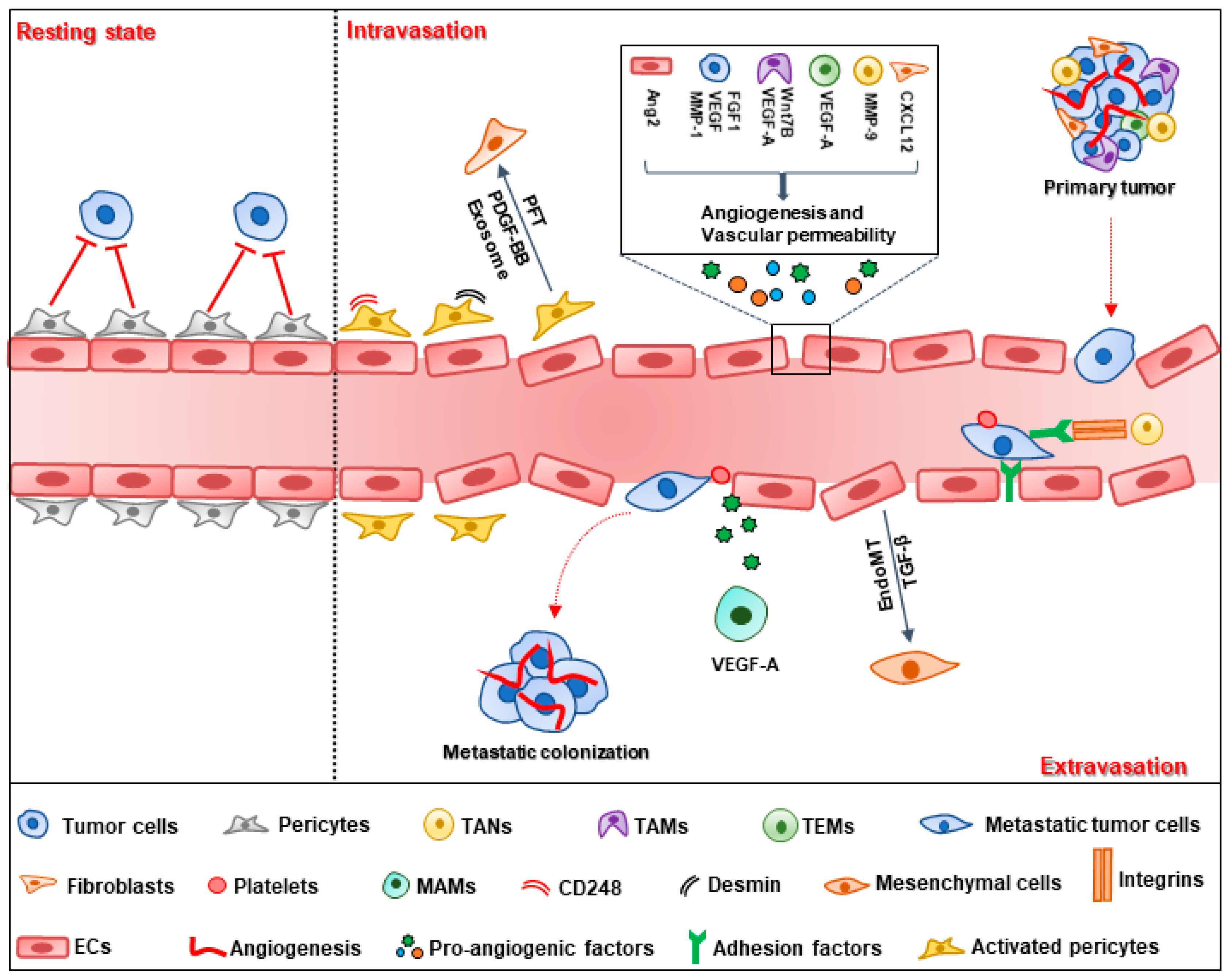
2.1. Tumor Cell Intravasation
2.2. Tumor Cell Extravasation
3. Application of Vessel Targeting Drugs in Metastatic Cancer
3.1. Monoclonal Antibody Therapy
3.2. Tyrosine Kinase Inhibitor Therapy
3.3. Fusion Protein Therapy
4. Mechanism of the Anti-Metastatic Effects Mediated by Vessel Targeting Therapies
4.1. Pre-Metastatic Niche Disruption
4.2. Vessel Normalization
5. Metastasis Risk after Vessel Targeting Therapies
5.1. EMT
5.2. Metabolism Shift
5.3. BMDC Recruitment
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paduch, R. The role of lymphangiogenesis and angiogenesis in tumor metastasis. Cell Oncol. (Dordr) 2016, 39, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.Y.; Hynes, R.O. Lymphatic or hematogenous dissemination: How does a metastatic tumor cell decide? Cell Cycle 2006, 5, 812–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kikuchi, H.; Ohta, M.; Kawabata, T.; Hiramatsu, Y.; Kondo, K.; Baba, M.; Kamiya, K.; Tanaka, T.; Kitagawa, M.; et al. TSU68 prevents liver metastasis of colon cancer xenografts by modulating the premetastatic niche. Cancer Res. 2008, 68, 9754–9762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, K.; Hu, J.; Korn, C.; Savant, S.; Teichert, M.; Kapel, S.S.; Jugold, M.; Besemfelder, E.; Thomas, M.; Pasparakis, M.; et al. Postsurgical adjuvant tumor therapy by combining anti-angiopoietin-2 and metronomic chemotherapy limits metastatic growth. Cancer Cell 2014, 26, 880–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- De Bock, K.; Mazzone, M.; Carmeliet, P. Antiangiogenic therapy, hypoxia, and metastasis: Risky liaisons, or not? Nat. Rev. Clin. Oncol. 2011, 8, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Nam, J.M.; Giaccia, A.J. Hypoxia: Signaling the Metastatic Cascade. Trends Cancer 2016, 2, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Mehlen, P.; Puisieux, A. Metastasis: A question of life or death. Nat. Rev. Cancer 2006, 6, 449–458. [Google Scholar] [CrossRef]
- Chiang, S.P.; Cabrera, R.M.; Segall, J.E. Tumor cell intravasation. Am. J. Physiol. Cell Physiol. 2016, 311, C1–C14. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, T.; Tsukikawa, S.; Yamada, K.; Yamaguchi, S. Morphologic analysis of microvessels in colorectal tumors with respect to the formation of liver metastases. J. Surg. Oncol. 2001, 78, 259–264. [Google Scholar] [CrossRef]
- Zhang, L.; Kharbanda, S.; McLeskey, S.W.; Kern, F.G. Overexpression of fibroblast growth factor 1 in MCF-7 breast cancer cells facilitates tumor cell dissemination but does not support the development of macrometastases in the lungs or lymph nodes. Cancer Res. 1999, 59, 5023–5029. [Google Scholar]
- Conn, E.M.; Botkjaer, K.A.; Kupriyanova, T.A.; Andreasen, P.A.; Deryugina, E.I.; Quigley, J.P. Comparative analysis of metastasis variants derived from human prostate carcinoma cells: Roles in intravasation of VEGF-mediated angiogenesis and uPA-mediated invasion. Am. J. Pathol. 2009, 175, 1638–1652. [Google Scholar] [CrossRef] [Green Version]
- Juncker-Jensen, A.; Deryugina, E.I.; Rimann, I.; Zajac, E.; Kupriyanova, T.A.; Engelholm, L.H.; Quigley, J.P. Tumor MMP-1 activates endothelial PAR1 to facilitate vascular intravasation and metastatic dissemination. Cancer Res. 2013, 73, 4196–4211. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wang, J.; Tholander, F.; Cao, Z.; Morikawa, H.; Tegner, J.; Yang, Y.; et al. VEGF-B promotes cancer metastasis through a VEGF-A-independent mechanism and serves as a marker of poor prognosis for cancer patients. Proc. Natl. Acad. Sci. USA 2015, 112, E2900–E2909. [Google Scholar] [CrossRef] [Green Version]
- Hakanpaa, L.; Sipila, T.; Leppanen, V.M.; Gautam, P.; Nurmi, H.; Jacquemet, G.; Eklund, L.; Ivaska, J.; Alitalo, K.; Saharinen, P. Endothelial destabilization by angiopoietin-2 via integrin beta1 activation. Nat. Commun. 2015, 6, 5962. [Google Scholar] [CrossRef]
- Bergers, G.; Song, S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massague, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.; Kim, J.; Cooke, V.G.; Wu, C.C.; Sugimoto, H.; Gu, C.; De Palma, M.; Kalluri, R.; LeBleu, V.S. Targeting vascular pericytes in hypoxic tumors increases lung metastasis via angiopoietin-2. Cell Rep. 2015, 10, 1066–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viski, C.; Konig, C.; Kijewska, M.; Mogler, C.; Isacke, C.M.; Augustin, H.G. Endosialin-Expressing Pericytes Promote Metastatic Dissemination. Cancer Res. 2016, 76, 5313–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, L.T.; Lockman, P.R.; Adkins, C.E.; Mohammad, A.S.; Sechrest, E.; Hua, E.; Palmieri, D.; Liewehr, D.J.; Steinberg, S.M.; Kloc, W.; et al. Alterations in Pericyte Subpopulations Are Associated with Elevated Blood-Tumor Barrier Permeability in Experimental Brain Metastasis of Breast Cancer. Clin. Cancer Res. 2016, 22, 5287–5299. [Google Scholar] [CrossRef] [Green Version]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [Green Version]
- Ning, X.; Zhang, H.; Wang, C.; Song, X. Exosomes Released by Gastric Cancer Cells Induce Transition of Pericytes Into Cancer-Associated Fibroblasts. Med. Sci. Monit. 2018, 24, 2350–2359. [Google Scholar] [CrossRef]
- Ahirwar, D.K.; Nasser, M.W.; Ouseph, M.M.; Elbaz, M.; Cuitino, M.C.; Kladney, R.D.; Varikuti, S.; Kaul, K.; Satoskar, A.R.; Ramaswamy, B.; et al. Fibroblast-derived CXCL12 promotes breast cancer metastasis by facilitating tumor cell intravasation. Oncogene 2018, 37, 4428–4442. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, E.Y.; Pollard, J.W. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007, 67, 5064–5066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeo, E.J.; Cassetta, L.; Qian, B.Z.; Lewkowich, I.; Li, J.F.; Stefater, J.A., 3rd; Smith, A.N.; Wiechmann, L.S.; Wang, Y.; Pollard, J.W.; et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014, 74, 2962–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Bekes, E.M.; Schweighofer, B.; Kupriyanova, T.A.; Zajac, E.; Ardi, V.C.; Quigley, J.P.; Deryugina, E.I. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am. J. Pathol. 2011, 179, 1455–1470. [Google Scholar] [CrossRef]
- Tien, Y.W.; Chang, K.J.; Jeng, Y.M.; Lee, P.H.; Wu, M.S.; Lin, J.T.; Hsu, S.M. Tumor angiogenesis and its possible role in intravasation of colorectal epithelial cells. Clin. Cancer Res. 2001, 7, 1627–1632. [Google Scholar]
- Strilic, B.; Offermanns, S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell. 2017, 32, 282–293. [Google Scholar] [CrossRef]
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870. [Google Scholar] [CrossRef]
- Chen, M.B.; Lamar, J.M.; Li, R.; Hynes, R.O.; Kamm, R.D. Elucidation of the Roles of Tumor Integrin beta1 in the Extravasation Stage of the Metastasis Cascade. Cancer Res. 2016, 76, 2513–2524. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Li, N.; Li, Z.; Chang, A.; Chen, Y.; Zhao, T.; Li, Y.; Wang, X.; Zhang, W.; Wang, Z.; et al. Tumour-derived Interleukin 35 promotes pancreatic ductal adenocarcinoma cell extravasation and metastasis by inducing ICAM1 expression. Nat. Commun. 2017, 8, 14035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strell, C.; Lang, K.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Surface molecules regulating rolling and adhesion to endothelium of neutrophil granulocytes and MDA-MB-468 breast carcinoma cells and their interaction. Cell Mol. Life Sci. 2007, 64, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Huh, S.J.; Liang, S.; Sharma, A.; Dong, C.; Robertson, G.P. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010, 70, 6071–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piera-Velazquez, S.; Li, Z.; Jimenez, S.A. Role of endothelial-mesenchymal transition (EndoMT) in the pathogenesis of fibrotic disorders. Am. J. Pathol. 2011, 179, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Krizbai, I.A.; Gasparics, A.; Nagyoszi, P.; Fazakas, C.; Molnar, J.; Wilhelm, I.; Bencs, R.; Rosivall, L.; Sebe, A. Endothelial-mesenchymal transition of brain endothelial cells: Possible role during metastatic extravasation. PLoS ONE 2015, 10, e0123845. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Al-Abd, A.M.; Alamoudi, A.J.; Abdel-Naim, A.B.; Neamatallah, T.A.; Ashour, O.M. Anti-angiogenic agents for the treatment of solid tumors: Potential pathways, therapy and current strategies—A review. J. Adv. Res. 2017, 8, 591–605. [Google Scholar] [CrossRef]
- De Falco, S. Antiangiogenesis therapy: An update after the first decade. Korean J. Intern. Med. 2014, 29, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hurwitz, H.; Fehrenbacher, L.; Novotny, W.; Cartwright, T.; Hainsworth, J.; Heim, W.; Berlin, J.; Baron, A.; Griffing, S.; Holmgren, E.; et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 2004, 350, 2335–2342. [Google Scholar] [CrossRef] [Green Version]
- Bennouna, J.; Sastre, J.; Arnold, D.; Osterlund, P.; Greil, R.; Van Cutsem, E.; von Moos, R.; Vieitez, J.M.; Bouche, O.; Borg, C.; et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): A randomised phase 3 trial. Lancet Oncol. 2013, 14, 29–37. [Google Scholar] [CrossRef]
- Loupakis, F.; Stein, A.; Ychou, M.; Hermann, F.; Salud, A.; Osterlund, P. A Review of Clinical Studies and Practical Guide for the Administration of Triplet Chemotherapy Regimens with Bevacizumab in First-line Metastatic Colorectal Cancer. Target. Oncol. 2016, 11, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Ilhan-Mutlu, A.; Osswald, M.; Liao, Y.; Gommel, M.; Reck, M.; Miles, D.; Mariani, P.; Gianni, L.; Lutiger, B.; Nendel, V.; et al. Bevacizumab Prevents Brain Metastases Formation in Lung Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudier, B.; Pluzanska, A.; Koralewski, P.; Ravaud, A.; Bracarda, S.; Szczylik, C.; Chevreau, C.; Filipek, M.; Melichar, B.; Bajetta, E.; et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet 2007, 370, 2103–2111. [Google Scholar] [CrossRef]
- Tewari, K.S.; Sill, M.W.; Long, H.J., 3rd; Penson, R.T.; Huang, H.; Ramondetta, L.M.; Landrum, L.M.; Oaknin, A.; Reid, T.J.; Leitao, M.M.; et al. Improved survival with bevacizumab in advanced cervical cancer. N. Engl. J. Med. 2014, 370, 734–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manso, L.; Moreno, F.; Marquez, R.; Castelo, B.; Arcediano, A.; Arroyo, M.; Ballesteros, A.I.; Calvo, I.; Echarri, M.J.; Enrech, S.; et al. Use of bevacizumab as a first-line treatment for metastatic breast cancer. Curr Oncol. 2015, 22, e51–e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero, A.J.; Escobar, M.; Lopes, G.; Gluck, S.; Vogel, C. Bevacizumab in the treatment of metastatic breast cancer: Friend or foe? Curr. Oncol. Rep. 2012, 14, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguerido, A.; Mulet-Margalef, N.; Matos, I.; Ros, J.; Argiles, G.; Elez, E.; Tabernero, J. The safety of ramucirumab for the treatment of colorectal cancer. Expert Opin. Drug Saf. 2018, 17, 945–951. [Google Scholar] [CrossRef]
- Arrieta, O.; Zatarain-Barron, Z.L.; Cardona, A.F.; Carmona, A.; Lopez-Mejia, M. Ramucirumab in the treatment of non-small cell lung cancer. Expert Opin. Drug Saf. 2017, 16, 637–644. [Google Scholar] [CrossRef]
- Aprile, G.; Rijavec, E.; Fontanella, C.; Rihawi, K.; Grossi, F. Ramucirumab: Preclinical research and clinical development. OncoTargets Ther. 2014, 7, 1997–2006. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Tagawa, S.T.; Kohli, M.; Eisen, A.; Canil, C.; Sridhar, S.S.; Spira, A.; Yu, E.Y.; Burke, J.M.; Shaffer, D.; et al. Docetaxel As Monotherapy or Combined With Ramucirumab or Icrucumab in Second-Line Treatment for Locally Advanced or Metastatic Urothelial Carcinoma: An Open-Label, Three-Arm, Randomized Controlled Phase II Trial. J. Clin. Oncol. 2016, 34, 1500–1509. [Google Scholar] [CrossRef]
- Ribatti, D.; Annese, T.; Ruggieri, S.; Tamma, R.; Crivellato, E. Limitations of Anti-Angiogenic Treatment of Tumors. Transl. Oncol. 2019, 12, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Reni, M.; Cereda, S.; Milella, M.; Novarino, A.; Passardi, A.; Mambrini, A.; Di Lucca, G.; Aprile, G.; Belli, C.; Danova, M.; et al. Maintenance sunitinib or observation in metastatic pancreatic adenocarcinoma: A phase II randomised trial. Eur. J. Cancer 2013, 49, 3609–3615. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.A.; Gore, M.E. Sunitinib in the treatment of metastatic renal cell carcinoma. Ther. Adv. Urol. 2016, 8, 348–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rini, B.I.; Hutson, T.E.; Figlin, R.A.; Lechuga, M.J.; Valota, O.; Serfass, L.; Rosbrook, B.; Motzer, R.J. Sunitinib in Patients With Metastatic Renal Cell Carcinoma: Clinical Outcome According to International Metastatic Renal Cell Carcinoma Database Consortium Risk Group. Clin. Genitourin Cancer 2018, 16, 298–304. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Yoshida, M.; Yamashita, T.; Okada, H.; Oishi, N.; Nio, K.; Hayashi, T.; Nomura, Y.; Hayashi, T.; Asahina, Y.; Ohwada, M.; et al. Sorafenib suppresses extrahepatic metastasis de novo in hepatocellular carcinoma through inhibition of mesenchymal cancer stem cells characterized by the expression of CD90. Sci. Rep. 2017, 7, 11292. [Google Scholar] [CrossRef]
- Jiang, C.; Xu, R.; Li, X.X.; Zhou, Y.F.; Xu, X.Y.; Yang, Y.; Wang, H.Y.; Zheng, X.F.S. Sorafenib and Carfilzomib Synergistically Inhibit the Proliferation, Survival, and Metastasis of Hepatocellular Carcinoma. Mol. Cancer Ther. 2018, 17, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schutz, G.; Thierauch, K.H.; Zopf, D. Regorafenib (BAY 73-4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2011, 129, 245–255. [Google Scholar] [CrossRef]
- Lyseng-Williamson, K.A. Regorafenib: A guide to its use in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib and sunitinib. BioDrugs 2013, 27, 525–531. [Google Scholar] [CrossRef]
- Duffaud, F.; Mir, O.; Boudou-Rouquette, P.; Piperno-Neumann, S.; Penel, N.; Bompas, E.; Delcambre, C.; Kalbacher, E.; Italiano, A.; Collard, O.; et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: A non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol. 2019, 20, 120–133. [Google Scholar] [CrossRef]
- Fallahi, P.; Ferrari, S.M.; Baldini, E.; Biricotti, M.; Ulisse, S.; Materazzi, G.; Miccoli, P.; Antonelli, A. The safety and efficacy of vandetanib in the treatment of progressive medullary thyroid cancer. Expert Rev. Anticancer Ther. 2016, 16, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Grullich, C. Cabozantinib: Multi-kinase Inhibitor of MET, AXL, RET, and VEGFR2. Recent Results Cancer Res. 2018, 211, 67–75. [Google Scholar] [PubMed]
- Smith, D.C.; Smith, M.R.; Sweeney, C.; Elfiky, A.A.; Logothetis, C.; Corn, P.G.; Vogelzang, N.J.; Small, E.J.; Harzstark, A.L.; Gordon, M.S.; et al. Cabozantinib in patients with advanced prostate cancer: Results of a phase II randomized discontinuation trial. J. Clin. Oncol. 2013, 31, 412–419. [Google Scholar] [CrossRef]
- Smith, M.; De Bono, J.; Sternberg, C.; Le Moulec, S.; Oudard, S.; De Giorgi, U.; Krainer, M.; Bergman, A.; Hoelzer, W.; De Wit, R.; et al. Phase III Study of Cabozantinib in Previously Treated Metastatic Castration-Resistant Prostate Cancer: COMET-1. J. Clin. Oncol. 2016, 34, 3005–3013. [Google Scholar] [CrossRef]
- Vecchio, S.J.D.; Ellis, R.J. Cabozantinib for the Management of Metastatic Clear Cell Renal Cell Carcinoma. J. Kidney Cancer VHL 2018, 5, 1–5. [Google Scholar] [CrossRef]
- Fala, L. Lenvima (Lenvatinib), a Multireceptor Tyrosine Kinase Inhibitor, Approved by the FDA for the Treatment of Patients with Differentiated Thyroid Cancer. Am. Health Drug Benefits 2015, 8, 176–179. [Google Scholar]
- Ikeda, K.; Kudo, M.; Kawazoe, S.; Osaki, Y.; Ikeda, M.; Okusaka, T.; Tamai, T.; Suzuki, T.; Hisai, T.; Hayato, S.; et al. Phase 2 study of lenvatinib in patients with advanced hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 512–519. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Okusaka, T.; Mitsunaga, S.; Ueno, H.; Tamai, T.; Suzuki, T.; Hayato, S.; Kadowaki, T.; Okita, K.; Kumada, H. Safety and Pharmacokinetics of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Sun, W. Ziv-aflibercept in metastatic colorectal cancer. Biologics 2014, 8, 13–25. [Google Scholar] [PubMed] [Green Version]
- Singh, S.R.; Dogra, A.; Stewart, M.; Das, T.; Chhablani, J. Intravitreal Ziv-Aflibercept: Clinical Effects and Economic Impact. Asia Pac. J. Ophthalmol (Phila) 2017, 6, 561–568. [Google Scholar]
- Ciombor, K.K.; Berlin, J.; Chan, E. Aflibercept. Clin. Cancer Res. 2013, 19, 1920–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, P.A.; Moore, M.J. Aflibercept in the treatment of patients with metastatic colorectal cancer: Latest findings and interpretations. Therap Adv. Gastroenterol. 2013, 6, 459–473. [Google Scholar] [CrossRef] [Green Version]
- Ellis, L.M. Bevacizumab. Nat. Rev. Drug Discov. 2005, 4, S8–S9. [Google Scholar] [CrossRef]
- Bennasroune, A.; Gardin, A.; Aunis, D.; Cremel, G.; Hubert, P. Tyrosine kinase receptors as attractive targets of cancer therapy. Crit. Rev. Oncol. Hematol. 2004, 50, 23–38. [Google Scholar] [CrossRef]
- Iyer, R.; Fetterly, G.; Lugade, A.; Thanavala, Y. Sorafenib: A clinical and pharmacologic review. Expert Opin. Pharmacother. 2010, 11, 1943–1955. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Matsui, J.; Matsushima, T.; Obaishi, H.; Miyazaki, K.; Nakamura, K.; Tohyama, O.; Semba, T.; Yamaguchi, A.; Hoshi, S.S.; et al. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc. Cell 2014, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 1992, 3, 65–71. [Google Scholar]
- Maj, E.; Papiernik, D.; Wietrzyk, J. Antiangiogenic cancer treatment: The great discovery and greater complexity (Review). Int. J. Oncol. 2016, 49, 1773–1784. [Google Scholar] [CrossRef] [Green Version]
- Marech, I.; Leporini, C.; Ammendola, M.; Porcelli, M.; Gadaleta, C.D.; Russo, E.; De Sarro, G.; Ranieri, G. Classical and non-classical proangiogenic factors as a target of antiangiogenic therapy in tumor microenvironment. Cancer Lett. 2016, 380, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Kanai, F.; Yoshida, H.; Tateishi, R.; Sato, S.; Kawabe, T.; Obi, S.; Kondo, Y.; Taniguchi, M.; Tagawa, K.; Ikeda, M.; et al. A phase I/II trial of the oral antiangiogenic agent TSU-68 in patients with advanced hepatocellular carcinoma. Cancer Chemother. Pharmacol. 2011, 67, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Viallard, C.; Larrivee, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 2017, 20, 409–426. [Google Scholar] [CrossRef]
- Korhonen, E.A.; Lampinen, A.; Giri, H.; Anisimov, A.; Kim, M.; Allen, B.; Fang, S.; D’Amico, G.; Sipila, T.J.; Lohela, M.; et al. Tie1 controls angiopoietin function in vascular remodeling and inflammation. J. Clin. Investig. 2016, 126, 3495–3510. [Google Scholar] [CrossRef] [Green Version]
- La Porta, S.; Roth, L.; Singhal, M.; Mogler, C.; Spegg, C.; Schieb, B.; Qu, X.; Adams, R.H.; Baldwin, H.S.; Savant, S.; et al. Endothelial Tie1-mediated angiogenesis and vascular abnormalization promote tumor progression and metastasis. J. Clin. Investig. 2018, 128, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2017, 31, 157–158. [Google Scholar] [CrossRef]
- Cantelmo, A.R.; Conradi, L.C.; Brajic, A.; Goveia, J.; Kalucka, J.; Pircher, A.; Chaturvedi, P.; Hol, J.; Thienpont, B.; Teuwen, L.A.; et al. Inhibition of the Glycolytic Activator PFKFB3 in Endothelium Induces Tumor Vessel Normalization, Impairs Metastasis, and Improves Chemotherapy. Cancer Cell 2016, 30, 968–985. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.B.; Tang, Y.L.; Liang, X.H. Targeting VEGF pathway to normalize the vasculature: An emerging insight in cancer therapy. OncoTargets Ther. 2018, 11, 6901–6909. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; Wong, A.H.; Jain, R.K. Vascular normalization as a therapeutic strategy for malignant and nonmalignant disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006486. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, M.; Oude Munnink, T.H.; Oosting, S.F.; Terwisscha van Scheltinga, A.G.; Gietema, J.A.; Garbacik, E.T.; Timmer-Bosscha, H.; Lub-de Hooge, M.N.; Schroder, C.P.; de Vries, E.G. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 2013, 73, 3347–3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K.; Duda, D.G.; Clark, J.W.; Loeffler, J.S. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat. Clin. Pract. Oncol. 2006, 3, 24–40. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.; Lee, L.Y.; Goh, K.Y.; Ong, R.; Hao, H.X.; Huang, A.; Wang, Y.; Graus Porta, D.; Chow, P.; Chung, A. Infigratinib Mediates Vascular Normalization, Impairs Metastasis, and Improves Chemotherapy in Hepatocellular Carcinoma. Hepatology 2019, 69, 943–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, A.S.; Kowanetz, M.; Wu, X.; Zhuang, G.; Ngu, H.; Finkle, D.; Komuves, L.; Peale, F.; Ferrara, N. Differential drug class-specific metastatic effects following treatment with a panel of angiogenesis inhibitors. J. Pathol. 2012, 227, 404–416. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Joseph, J.P.; Harishankar, M.K.; Pillai, A.A.; Devi, A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018, 80, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J. Clin. Med. 2016, 5, 41. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhang, C.; Cui, M.; Niu, J.; Ding, W. Inhibition of Bevacizumab-induced Epithelial-Mesenchymal Transition by BATF2 Overexpression Involves the Suppression of Wnt/beta-Catenin Signaling in Glioblastoma Cells. Anticancer Res. 2017, 37, 4285–4294. [Google Scholar] [PubMed]
- Zhang, W.; Sun, H.C.; Wang, W.Q.; Zhang, Q.B.; Zhuang, P.Y.; Xiong, Y.Q.; Zhu, X.D.; Xu, H.X.; Kong, L.Q.; Wu, W.Z.; et al. Sorafenib down-regulates expression of HTATIP2 to promote invasiveness and metastasis of orthotopic hepatocellular carcinoma tumors in mice. Gastroenterology 2012, 143, 1641–1649.e5. [Google Scholar] [CrossRef]
- Keunen, O.; Johansson, M.; Oudin, A.; Sanzey, M.; Rahim, S.A.; Fack, F.; Thorsen, F.; Taxt, T.; Bartos, M.; Jirik, R.; et al. Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3749–3754. [Google Scholar] [CrossRef] [Green Version]
- Kumar, K.; Wigfield, S.; Gee, H.E.; Devlin, C.M.; Singleton, D.; Li, J.L.; Buffa, F.; Huffman, M.; Sinn, A.L.; Silver, J.; et al. Dichloroacetate reverses the hypoxic adaptation to bevacizumab and enhances its antitumor effects in mouse xenografts. J. Mol. Med. (Berl) 2013, 91, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.L.; et al. Fatty acid uptake and lipid storage induced by HIF-1alpha contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [Green Version]
- Papaevangelou, E.; Almeida, G.S.; Box, C.; deSouza, N.M.; Chung, Y.L. The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int J. Cancer 2018, 143, 992–1002. [Google Scholar] [CrossRef]
- Sounni, N.E.; Cimino, J.; Blacher, S.; Primac, I.; Truong, A.; Mazzucchelli, G.; Paye, A.; Calligaris, D.; Debois, D.; De Tullio, P.; et al. Blocking lipid synthesis overcomes tumor regrowth and metastasis after antiangiogenic therapy withdrawal. Cell Metab. 2014, 20, 280–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McIntyre, A.; Harris, A.L. Metabolic and hypoxic adaptation to anti-angiogenic therapy: A target for induced essentiality. EMBO Mol. Med. 2015, 7, 368–379. [Google Scholar] [CrossRef] [PubMed]
- Achyut, B.R.; Arbab, A.S. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. OncoTargets Ther. 2016, 9, 1047–1055. [Google Scholar] [PubMed] [Green Version]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.Y.; Gordon-Weeks, A.; Allen, D.; Kersemans, V.; Beech, J.; Smart, S.; Muschel, R.J. Cd11b(+) myeloid cells support hepatic metastasis through down-regulation of angiopoietin-like 7 in cancer cells. Hepatology 2015, 62, 521–533. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.Y.; Yuen, V.W.; Wong, C.C. Hypoxia and the Metastatic Niche. Adv. Exp. Med. Biol. 2019, 1136, 97–112. [Google Scholar]
- Dost Gunay, F.S.; Kirmizi, B.A.; Ensari, A.; Icli, F.; Akbulut, H. Tumor-associated Macrophages and Neuroendocrine Differentiation Decrease the Efficacy of Bevacizumab Plus Chemotherapy in Patients With Advanced Colorectal Cancer. Clin. Colorectal Cancer 2019, 18, e244–e250. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Stintzing, S.; Cao, S.; Heinemann, V.; Cremolini, C.; Falcone, A.; Yang, D.; Zhang, W.; Ning, Y.; Stremitzer, S.; et al. Variations in genes regulating tumor-associated macrophages (TAMs) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: Results from TRIBE and FIRE3 trials. Ann. Oncol. 2015, 26, 2450–2456. [Google Scholar]
- Zhang, W.; Zhu, X.D.; Sun, H.C.; Xiong, Y.Q.; Zhuang, P.Y.; Xu, H.X.; Kong, L.Q.; Wang, L.; Wu, W.Z.; Tang, Z.Y. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010, 16, 3420–3430. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Yang, Y.; Lu, M.; Lei, Y.; Xu, L.; Jiang, Z.; Xu, X.; Guo, X.; Zhang, X.; Sun, H.; et al. Recent Advances in the Discovery of HIF-1alpha-p300/CBP Inhibitors as Anti-Cancer Agents. Mini Rev. Med. Chem. 2018, 18, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Saez, O.; Gajate Borau, P.; Alonso-Gordoa, T.; Molina-Cerrillo, J.; Grande, E. Targeting HIF-2 alpha in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit. Rev. Oncol. Hematol. 2017, 111, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.V.; Williams, K.J.; Cowen, R.L.; Jaffar, M.; Telfer, B.A.; Saunders, M.; Airley, R.; Honess, D.; van der Kogel, A.J.; Wolf, C.R.; et al. Oxygen-sensitive enzyme-prodrug gene therapy for the eradication of radiation-resistant solid tumours. Gene Ther. 2002, 9, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Silva, V.L.; Al-Jamal, W.T. Exploiting the cancer niche: Tumor-associated macrophages and hypoxia as promising synergistic targets for nano-based therapy. J. Control. Release 2017, 253, 82–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guise, C.P.; Mowday, A.M.; Ashoorzadeh, A.; Yuan, R.; Lin, W.H.; Wu, D.H.; Smaill, J.B.; Patterson, A.V.; Ding, K. Bioreductive prodrugs as cancer therapeutics: Targeting tumor hypoxia. Chin. J. Cancer 2014, 33, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Horton, M.R.; Powell, J.D. Something in the air: Hyperoxic conditioning of the tumor microenvironment for enhanced immunotherapy. Cancer Cell 2015, 27, 435–436. [Google Scholar] [CrossRef] [Green Version]
- Casazza, A.; Laoui, D.; Wenes, M.; Rizzolio, S.; Bassani, N.; Mambretti, M.; Deschoemaeker, S.; Van Ginderachter, J.A.; Tamagnone, L.; Mazzone, M. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell 2013, 24, 695–709. [Google Scholar] [CrossRef] [Green Version]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Johansson, A.; Hamzah, J.; Ganss, R. More than a scaffold: Stromal modulation of tumor immunity. Biochim. Biophys. Acta 2016, 1865, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terme, M.; Pernot, S.; Marcheteau, E.; Sandoval, F.; Benhamouda, N.; Colussi, O.; Dubreuil, O.; Carpentier, A.F.; Tartour, E.; Taieb, J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013, 73, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittnaegel, M.; Rigamonti, N.; Kadioglu, E.; Cassara, A.; Wyser Rmili, C.; Kiialainen, A.; Kienast, Y.; Mueller, H.J.; Ooi, C.H.; Laoui, D.; et al. Dual angiopoietin-2 and VEGFA inhibition elicits antitumor immunity that is enhanced by PD-1 checkpoint blockade. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Di Tacchio, M.; Macas, J.; Weissenberger, J.; Sommer, K.; Bahr, O.; Steinbach, J.P.; Senft, C.; Seifert, V.; Glas, M.; Herrlinger, U.; et al. Tumor Vessel Normalization, Immunostimulatory Reprogramming, and Improved Survival in Glioblastoma with Combined Inhibition of PD-1, Angiopoietin-2, and VEGF. Cancer Immunol. Res. 2019. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Nadal, R.; Amin, A.; Geynisman, D.M.; Voss, M.H.; Weinstock, M.; Doyle, J.; Zhang, Z.; Viudez, A.; Plimack, E.R.; McDermott, D.F.; et al. Safety and clinical activity of vascular endothelial growth factor receptor (VEGFR)-tyrosine kinase inhibitors after programmed cell death 1 inhibitor treatment in patients with metastatic clear cell renal cell carcinoma. Ann. Oncol. 2016, 27, 1304–1311. [Google Scholar] [CrossRef]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Donnem, T.; Hu, J.; Ferguson, M.; Adighibe, O.; Snell, C.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Vessel co-option in primary human tumors and metastases: An obstacle to effective anti-angiogenic treatment? Cancer Med. 2013, 2, 427–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518–529. [Google Scholar] [CrossRef]
- Frentzas, S.; Simoneau, E.; Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Kostaras, E.; Nathan, M.; Wotherspoon, A.; Gao, Z.H.; Shi, Y.; et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat. Med. 2016, 22, 1294–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bridgeman, V.L.; Vermeulen, P.B.; Foo, S.; Bilecz, A.; Daley, F.; Kostaras, E.; Nathan, M.R.; Wan, E.; Frentzas, S.; Schweiger, T.; et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J. Pathol. 2017, 241, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Leenders, W.P.; Kusters, B.; Verrijp, K.; Maass, C.; Wesseling, P.; Heerschap, A.; Ruiter, D.; Ryan, A.; de Waal, R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin. Cancer Res. 2004, 10, 6222–6230. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agent | Type of Inhibitor | Targets | Clinical Application |
|---|---|---|---|
| Bevacizumab | Monoclonal antibody | VEGF-A | Metastatic colorectal cancer [48,49,50,51] Metastatic nonsquamous non-small cell lung cancer [52] Metastatic renal cell carcinoma [53] Metastatic cervical cancer [54] metastatic breast cancer [55,56] |
| Ramucirumab | Monoclonal antibody | VEGFR-2 | Metastatic colorectal cancer [57] Metastatic non-small cell lung cancer [58] Metastatic stomach adenocarcinoma [59] Metastatic gastroesophageal junction adenocarcinoma [59] Metastatic urothelial carcinoma [60] |
| Sunitinib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -2, VEGFR -3, PDGFR-α, PDGFR-β, c-Kit, CSF-1R, RET | Metastatic pancreatic cancer [61,62] Metastatic renal cell carcinoma [63,64]. |
| Sorafenib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -2, VEGFR -3, PDGFR-β, Flt-3, c-Kit | Metastatic hepatocellular carcinoma [65,66,67] Metastatic thyroid cancer [68] |
| Regorafenib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR -3, PDGFR-β, FGFR-1 KIT, RET and B-RAF | Metastatic colorectal cancer [69] Metastatic gastrointestinal stromal tumor [70] Metastatic osteosarcoma [71] |
| Vandetanib | Tyrosine kinase inhibitor | VEGFR-2, VEGFR-3, EGFR, RET | Metastatic medullary thyroid cancer [72] |
| Cabozantinib | Tyrosine kinase inhibitor | MET, RET, AXL, VEGFR-2, FLT3, c-Kit | Metastatic medullary thyroid cancer [73] Metastatic castration-resistant prostate cancer [74,75] Metastatic renal cell carcinoma [76] |
| Lenvatinib | Tyrosine kinase inhibitor | VEGFR-1, VEGFR-2, VEGFR-3 FGFR, PDGFRα, KIT and RET | Metastatic differentiated thyroid cancer [77] Metastatic hepatocellular carcinoma [78,79] Metastatic renal cell carcinoma [80] |
| Aflibercept | Fusion protein | VEGF-A, VEGF-B, PlGF | Metastatic colorectal cancer [81,82,83,84] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Li, Y.; Lu, W.; Chen, M.; Ye, W.; Zhang, D. The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis. Cells 2019, 8, 1602. https://doi.org/10.3390/cells8121602
Li X, Li Y, Lu W, Chen M, Ye W, Zhang D. The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis. Cells. 2019; 8(12):1602. https://doi.org/10.3390/cells8121602
Chicago/Turabian StyleLi, Xiaobo, Yong Li, Weijin Lu, Minfeng Chen, Wencai Ye, and Dongmei Zhang. 2019. "The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis" Cells 8, no. 12: 1602. https://doi.org/10.3390/cells8121602
APA StyleLi, X., Li, Y., Lu, W., Chen, M., Ye, W., & Zhang, D. (2019). The Tumor Vessel Targeting Strategy: A Double-Edged Sword in Tumor Metastasis. Cells, 8(12), 1602. https://doi.org/10.3390/cells8121602