1. Introduction
According to the World Health Organization, the leading causes of death worldwide are stroke (ST), chronic obstructive pulmonary disease (COPD), Alzheimer’s disease (AD), and tuberculosis (TB) [
1]. ST has been shown to have a higher incidence in developing countries than in developed countries. Of the various types of ST, 85% are ischemic. It has also been suggested that both genetic and environmental factors are involved in pathophysiology [
2]. COPD is caused by an inflammatory response in the lungs to harmful gas or particles. COPD is progressive and often coexists with other diseases. The most common risk factor is smoking [
3]. AD, the most common cause of dementia, is a progressive neurodegenerative disease that develops due to neuronal death and loss or atrophy of synaptic functions [
4]. TB is an infectious disease caused by
Mycobacterium tuberculosis that causes death beyond AIDS due to HIV. Currently, it is estimated that one-third of the world’s population is potentially infected. However, it affects about 5–10% of infected people [
5].
MicroRNA (miRNA) is attracting attention as a treatment method and biomarker for these diseases [
6]. miRNA is a class of small non-coding RNA, containing around 22 nucleotides. miRNA mainly controls the turnover and translation of target mRNA by binding the 3’ untranslated region of mRNA as a target site [
7]. Currently, seed theory is the mainstream in binding miRNA targets [
8]. Seed theory states that the 2nd to 8th bases of miRNA bind complementarily to the target site. Various target prediction programs have been developed based on this theory, but it is known that they often have high false positive rates or false negative rates [
9]. miRNAs are related to various biological processes because of their functions, and their relation to diseases is well studied [
10,
11]. In order to clarify the control mechanism of biological process by miRNA, a new theory for the miRNA mechanism may be necessary. For this reason, several studies have recently been conducted to clarify the relationship between diseases and miRNAs using methods that are not based on seed theory. Zou et al. combined the miRNA–miRNA network, miRNA–disease network, and disease–disease network, and performed network analysis by machine learning [
12]. Luo et al., Zou et al., Lan et al., and Fu et al. applied network analysis by machine learning to miRNA–disease networks [
9,
13,
14,
15]. Ding et al. focused on miRNA clusters/family and secondary structure [
16], and Zheng et al. focused on miRNA’s functional similarity and semantic similarity to diseases [
17]. These methods can be expected to enable prediction of disease-related miRNA candidates that have not yet been found. For example,
hsa –let –7d,
hsa –mir –18a,
hsa –mir –145,
hsa –mir –106b,
hsa –let –7e,
hsa –let –7b,
hsa –mir –19a, and
hsa –mir –125a may be related to breast cancer [
12].
Seed theory may be superior in terms of the mechanism by which miRNA suppresses translation of mRNA and the prediction of miRNA-mRNA networks. In addition, from the viewpoint of predicting disease-related miRNA candidates that have not yet been found, the above-described approach using machine learning is considered to be excellent. However, neither method may be suitable in terms of decoding the miRNA gene. This is because seed theory focuses on sequence specificity when miRNA is viewed as a translation inhibitor of mRNA, and machine learning approaches are not based on sequences. Decoding miRNA genes not only reveals the mechanism of miRNA-mRNA interactions and miRNA-non-coding RNA interactions, but may also reveal the role of short RNAs in primitive life, such as in its birth and evolution.
In order to clarify the relationship between biological phenomena and miRNA sequences, the concept of black box modeling in system identification in the engineering field was used. In black box modeling, an internal system is regarded as a black box, and system identification is performed using only Input and Output. In this study, Input was designated as miRNA sequence and Output as disease. The RNA-RNA interaction was the focus of attention when quantifying miRNA sequences. The purpose of this study was to verify whether this new approach reveals the relationship between miRNA and ST, COPD, AD, and TB.
4. Discussion
Since miRNA is deeply involved in various biological reactions including diseases, it is important to know the mechanism in detail in the treatment of diseases [
20,
21]. The mechanism by which miRNA suppresses translation has been clarified [
22], but the meaning of the sequence when miRNA is regarded as a gene has not yet been clarified. The seed region is considered important in the seed theory, but the array outside the seed region has not been fully elucidated. Even in the seed region, the relationship with the disease is unclear in terms of the base sequence. Elucidating the relationship between miRNA sequences and diseases is the first step to deciphering miRNA genes. However, it seems that conventional methods are difficult in terms of decoding miRNA genes. Therefore, we focused on RNA–RNA interactions. Nucleobase hydrogen bonds are utilized when reacting with other nucleic acids. Therefore, the base sequence was digitized by using AUHB as the miRNA score, and the relationship with the disease was verified.
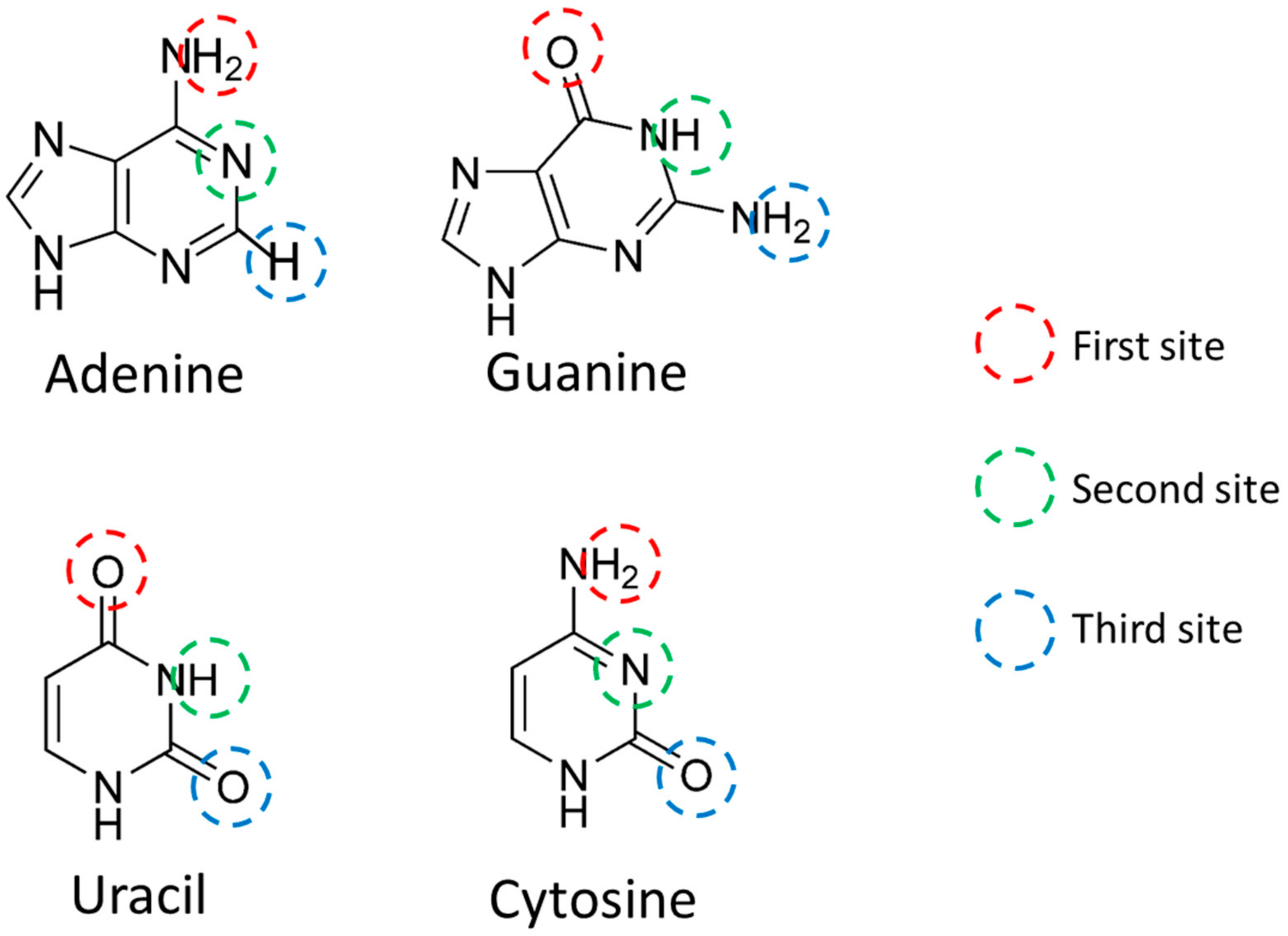
In order to quantify the miRNA sequence, the charge was calculated focusing on AUHB (
Table 1). The H was a positive value, and O and nitrogen atoms were negative values. In the Watson–Crick pair, the third site of A does not bond with hydrogen, and in this charge calculation, only the charge of the third site of A was smaller in absolute value than other atoms. Therefore, this calculation result was interpreted as appropriate.
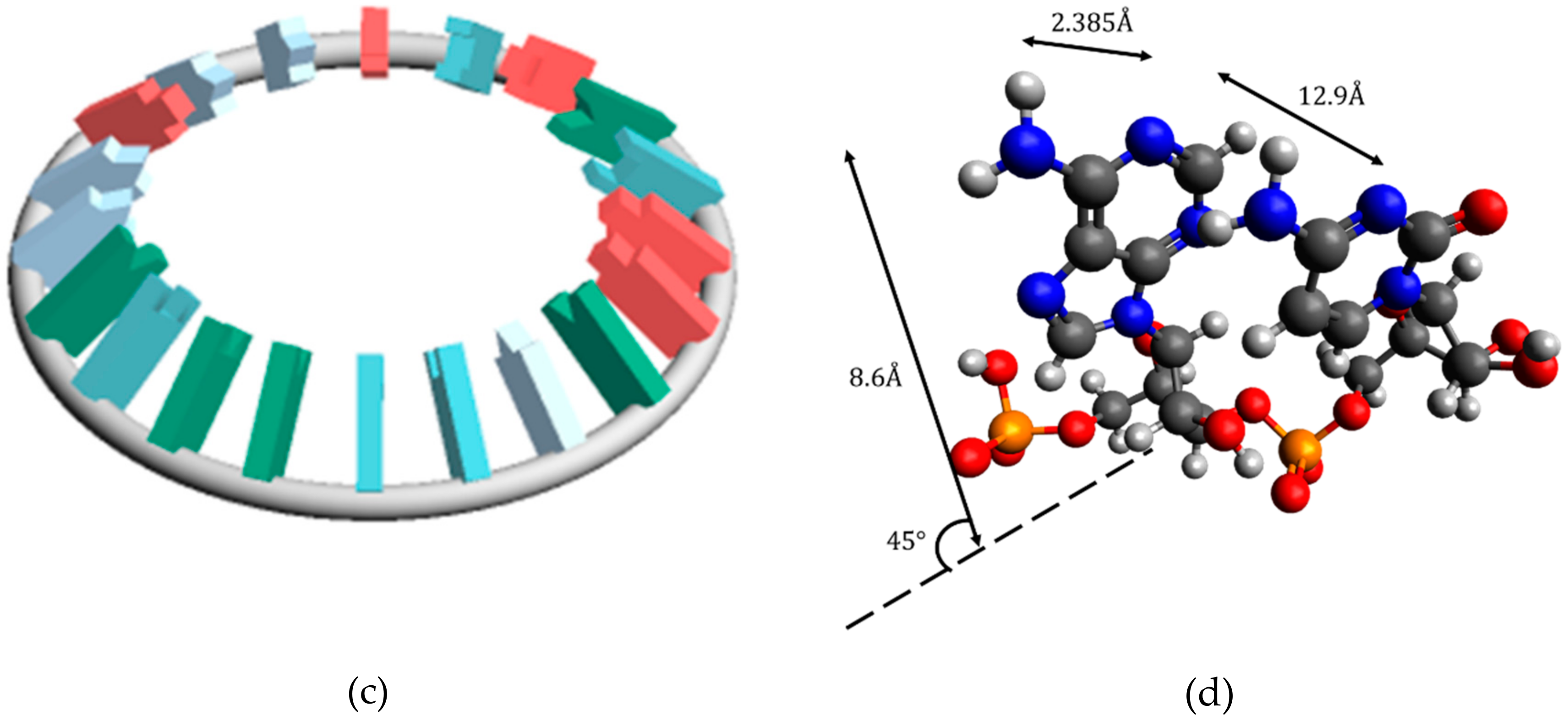
In this study, miRNA was considered a torus. There are two reasons for this. First of all, it is known that there are circular forms of various RNA types. For example, circular RNAs have been reported [
23,
24]. In addition, tRNA is clover-shaped and can be regarded as circular in terms of topology. Pre-mRNA [
25] and phytopathogenic virus viroid [
26] are also circular. Although we have not found any miRNA reports that are circular, miRNA secondary structure suggests a circular shape. For example, the secondary structure of mature miRNA can be predicted with CentroidFold [
27]. When predicting the secondary structure of miRNA in CentroidFold, miRNA basically has a dumbbell shape, but some miRNAs become torus (e.g., hsa-miR-1-5p; inference engine: CONTRAfold; weight of base pairs: 2
2). The dumbbell type is also topologically similar to a torus. Second, considering miRNA as a torus has two advantages. The first is that the clover type and dumbbell type can be considered topologically homologous, so the method of this study may be applicable to other types of RNA. Secondly, the readability of the miRNA genetic code sequence is high. In order to decipher the genetic code of miRNA sequences as the ultimate goal, this study aims to search for pre-data processing methods for machine learning. It is probably difficult to perform data preprocessing because the amount of information is too small for a one-dimensional array. On the other hand, there is too much information in an accurate secondary structure or tertiary structure. A torus can be viewed as an approximation of secondary structure.
Based on the calculation results, each
was calculated. In order to clarify the relationship between miRNA sequences and diseases, it is desirable that
and sequences have a 1: 1 relationship. Therefore, the ratio of unique values in each
was verified (
Table 2). The EV_C type and EV_S type differ greatly in the degree of overlap, and the EV_C type had more overlap. This is thought to be because the EV_C type is a vector with only the centre, so the influence from the charge is small, whereas the EV_S type is the sum of the electric field vectors in the torus, so it is strongly influenced by the charge. In addition, the EV_C/S type had more overlap in structure A than in others. This is presumably because the z component of the electric field vector does not exist. Surprisingly, Sum had a low overlap rate compared to the EV_C type and EV_S_A. From the viewpoint of unique values, Sum, EV_S_B, and EV_S_C were considered suitable.
A single regression analysis was performed to examine the distribution trend of each
value (
Table 3). Overall correlation was high except for Sum.
is a value uniquely determined by the nucleobase, but
itself did not show similarities in the values of each element except for the values of the second and third sites of G. Nevertheless, it was unexpected that the correlation was high. Note that the strong correlation in the order of VS, EV_S, EV_C, and Sum is thought to be due to the difference in strength affected by
. From the viewpoint of correlation, VS and EV_S types were considered suitable.
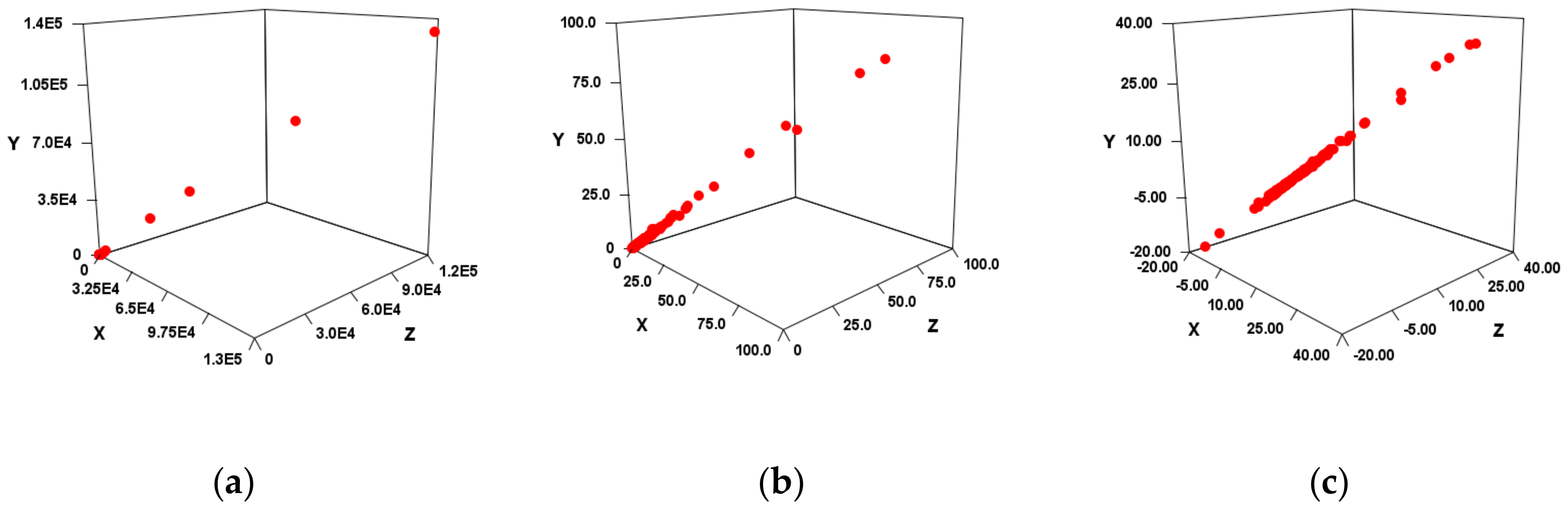
Since the correlation was high in each
, it was suggested that SMEL could also have a high correlation. When tested in four diseases, VS and EV_S types showed a high correlation in SMEL, and EV_C type and Sum showed low correlation (
Figure 3,
Table 4). As described above, since the SMEL intercept is 0, the regression line of each disease is non–parallel. If there is no relationship between the miRNA sequence and the disease, it should be almost the same as the slope in
. However, there was actually a significant difference in the slope (
Table 3 and
Table 4). In other words, the high correlation of SMEL is thought to be due to the correlation between the disease and miRNA, although it is influenced by the high correlation of
.
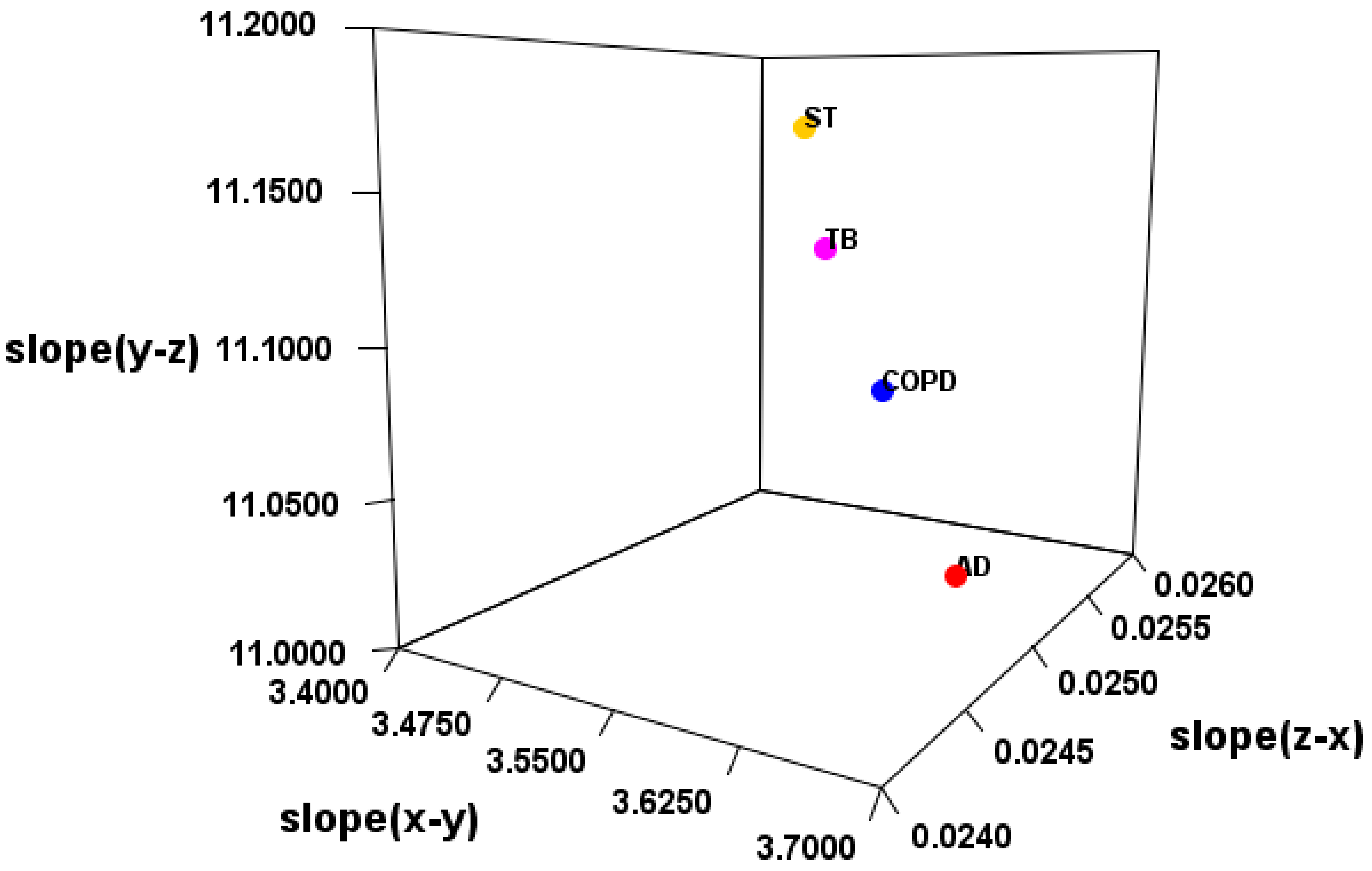
Therefore, it was verified whether the disease can be distinguished by SMEL (
Table 5 and
Table 6). As a result, EV_C_A, EV_S_B, or EV_S_C was considered a suitable method. However, there may be large outliers in the case of FC (
Figure 3a,b). Since these are linear regressions, outliers can have a significant impact in the case of FC. Therefore, it was verified even when outliers were not taken into account (
Table 7). Based on the results with and without outliers, the most suitable
was considered to be EV_S_B.
EV_S_B was suitable for unique values, correlations, and SMEL. The reason why EV_S_C, which showed the same tendency as EV_S_B, did not show a significant difference at 2σ of SMEL, seems to be because EV_S_B was closest to the possible structure.
It was suggested that the classification of the disease is possible by the electric field vector made by AUHB (
Figure 4), but there are three main problems in this result. The first is that the structure is simplified. In the case of EV_S_B, which seemed most suitable, the First to Third sites were placed perpendicular to the plane of the torus (
Figure 2b). However, the actual nucleobase of miRNA is not vertical, but seems to be angled by the π–π interaction and the phosphate–sugar chain interaction. Therefore, it is necessary to perform calculations in consideration of the angle. The second is that only four types of disease are compared. Thus, in order to prove the hypothesis that “the relationship between the miRNA sequence and disease may be revealed by focusing on hydrogen bonding sites in RNA–RNA interaction”, more diseases must be targeted. Furthermore, in this study, comparisons were made between different diseases in major classifications, but it is also necessary to verify that diseases with the same large category, such as cancer types, can be distinguished. The third is interpretation of the score. Since EV_S_B is almost sequence-specific, the results of this study are considered to be one step closer to miRNA gene decoding. However, as mentioned in the second problem, there are still only few subjects tested in the experiment. Therefore, it is not possible at this time to interpret the score. In order to interpret the score, it is necessary to collect a lot of data and perform machine learning. Therefore, it is necessary to continue research in the future.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}