A High Efficient Biological Language Model for Predicting Protein–Protein Interactions

Abstract
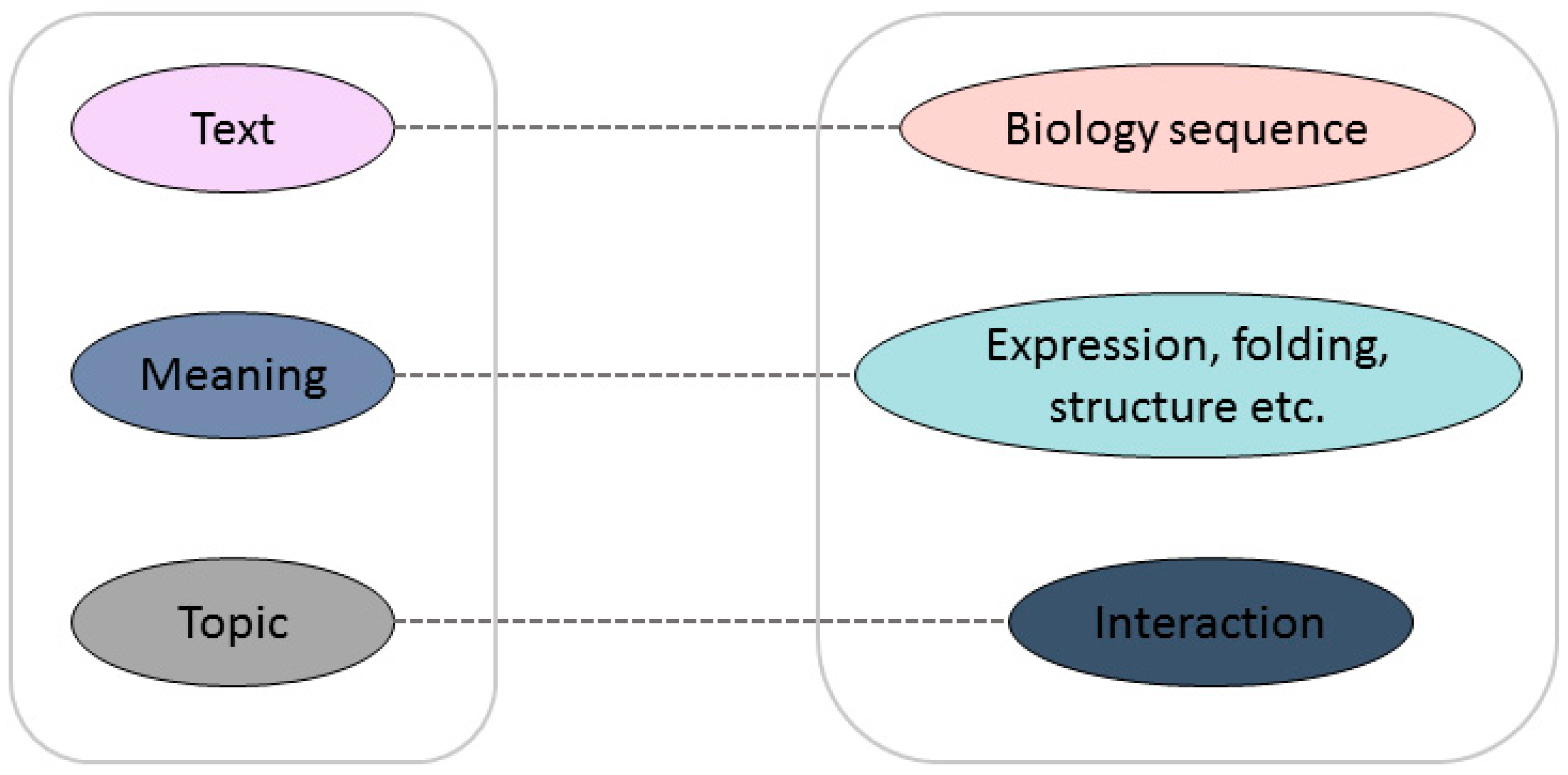
:1. Introduction
2. Materials and Methods
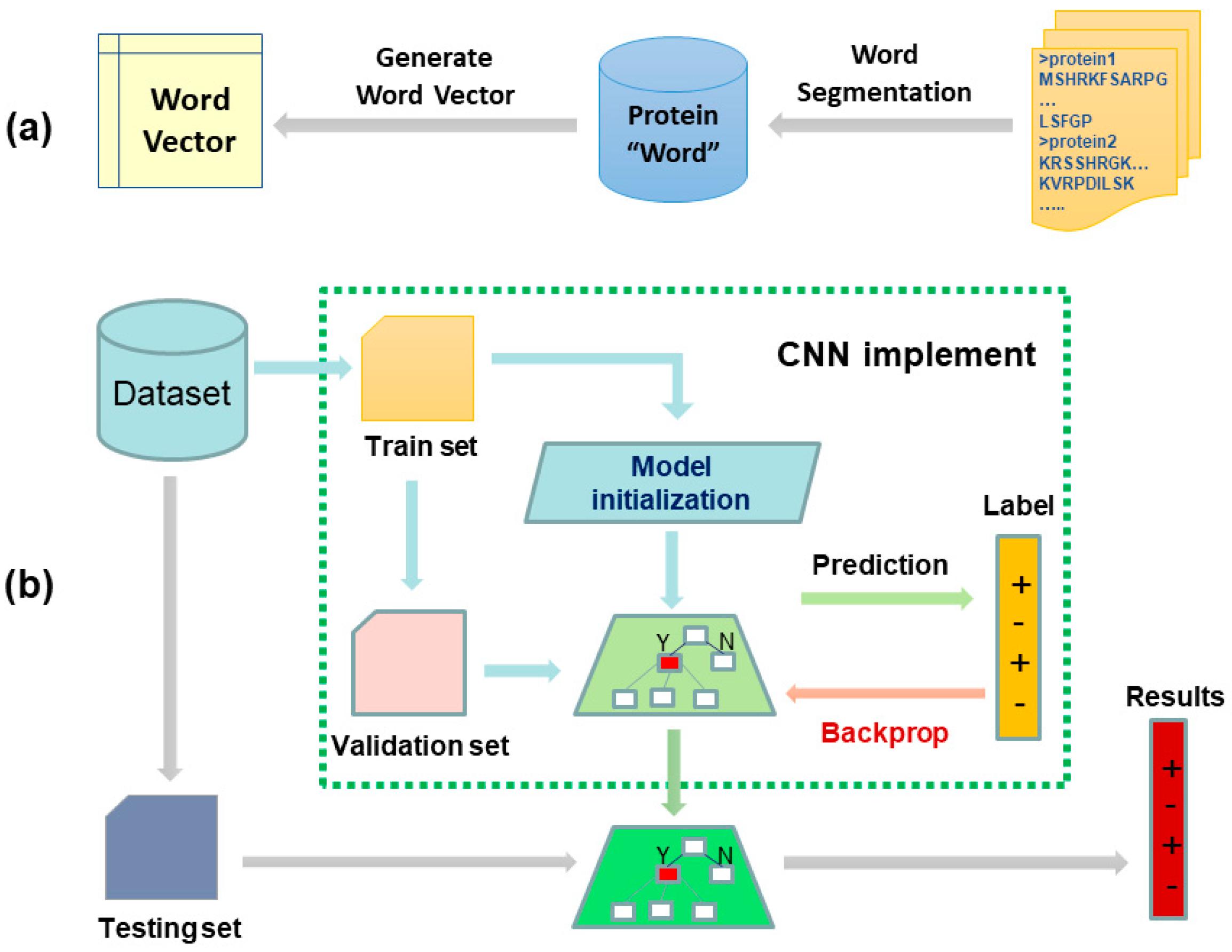
2.1. Data Construction
2.2. Bio-Word Segmentation
- We created a reasonable seed vocabulary using the union of all amino acids and the most frequent “amino acid string” in our protein sequence data set. Here, Byte-Pair-Encoding (BPE) algorithm [20] can be used to conduct this step. The BPE first split every protein sequence into individual amino acids. The most frequent adjacent pairs of amino acids were then consecutively merged until reaching a desired seed vocabulary size. Frequent “amino acid string” can be enumerated by the Enhanced Suffix Array algorithm [21], which only takes O(T) time and O(20T) space.
- Repeat the following steps until Φ reaches a desired vocabulary size.
- (a)
- Fixing the set of vocabulary, optimize by adopting the EM algorithm.
- (b)
- For each word , we computed the that measured the change of likelihood , when the word was removed from the current vocabulary.
- (c)
- Sort words based on loss and keep top α% (α is 70, in this paper).
2.3. Feature Extraction
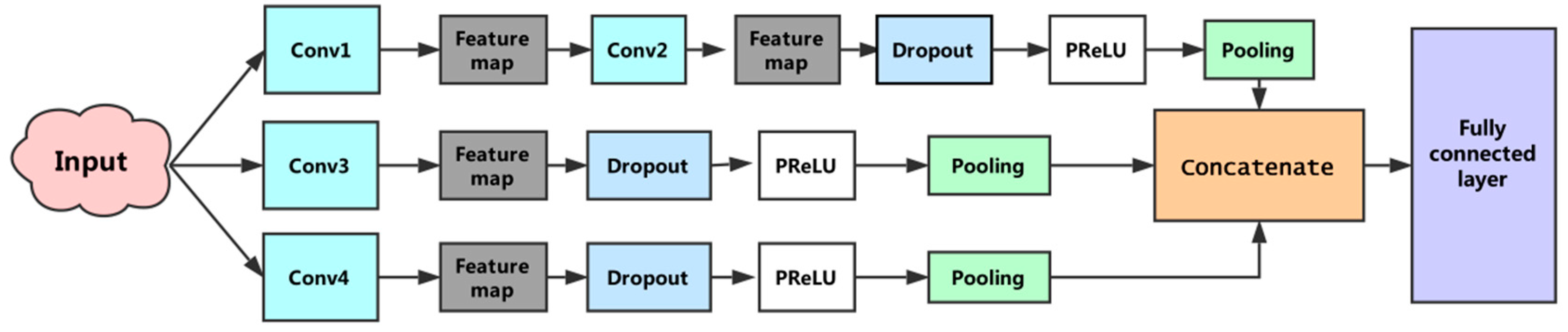
2.4. CNN Construction
3. Results
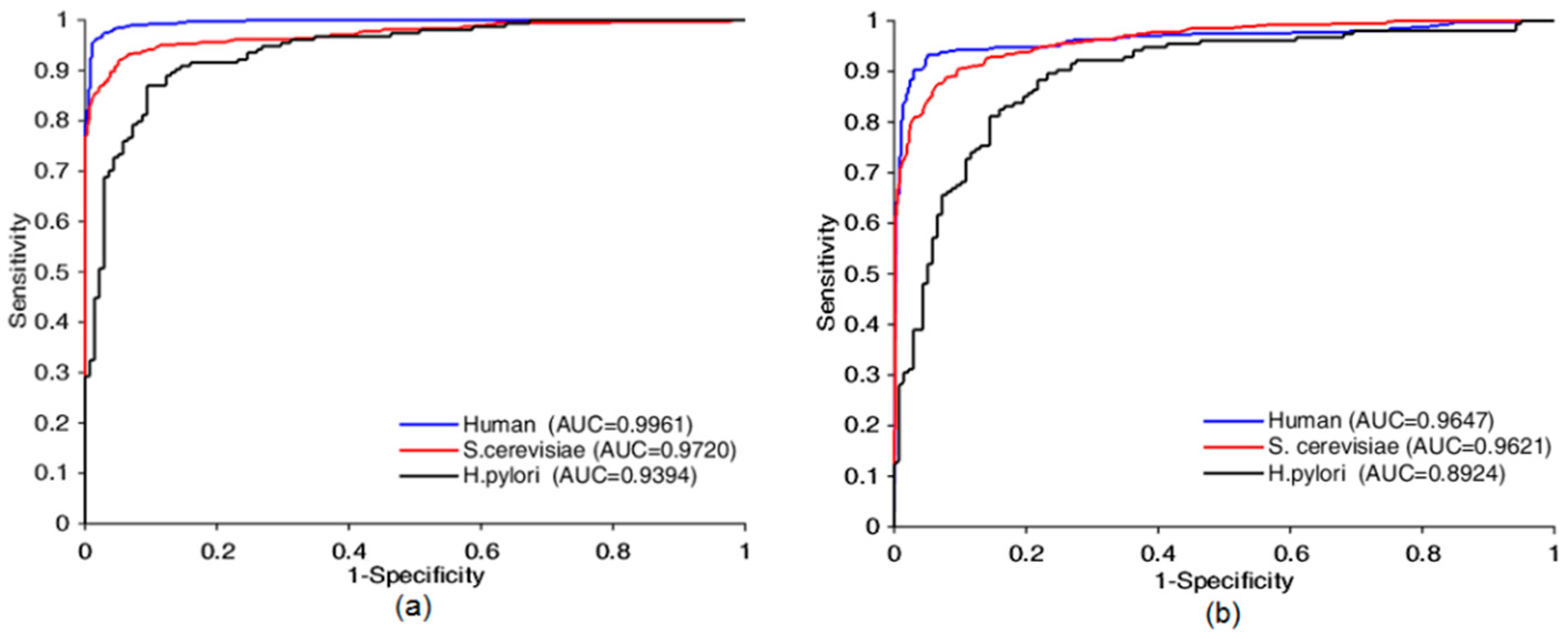
3.1. Prediction Performances on Three PPIs Data Sets
3.2. Comparison of Different Word Segmentation Schemes
3.3. Comparison with Previous Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability
References
- Koegl, M.; Uetz, P. Improving yeast two-hybrid screening systems. Brief Funct. Genom. Proteom. 2007, 6, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, N.; Sakakibara, Y. Statistical prediction of protein–chemical interactions based on chemical structure and mass spectrometry data. Bioinformatics 2007, 23, 2004–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rüetschi, U.; Rosén, A.; Karlsson, G.; Zetterberg, H.; Rymo, L.; Hagberg, H.; Jacobsson, B. Proteomic analysis using protein chips to detect biomarkers in cervical and amniotic fluid in women with intra-amniotic inflammation. J. Proteome Res. 1900, 4, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yamanishi, Y.; Kanehisa, M.; Toh, H.; Jp, U.A.; Kyoto, T.K. Prediction of protein-protein interactions based on real-valued phylogenetic profiles using partial correlation coefficient. BMC Genom. 2004, 10, 288. [Google Scholar]
- Huang, C.; Morcos, F.; Kanaan, S.P.; Wuchty, S.; Chen, D.Z.; Izaguirre, J.A. Predicting protein-protein interactions from protein domains using a set cover approach. IEEE/ACM Trans. Comput. Biol. Bioinform. 2007, 4, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Han, J.J.; Dupuy, D.; Bertin, N.; Cusick, M.E.; Vidal, M. Effect of sampling on topology predictions of protein-protein interaction networks. Nat. Biotechnol. 2005, 23, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.C.; Cai, Y.D. Predicting protein-protein interactions from sequences in a hybridization space. J. Proteome Res. 2006, 5, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, J.; Luo, X.; Zhu, W.; Yu, K.; Chen, K.; Li, Y.; Jiang, H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA 2007, 104, 4337–4341. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Yu, L.; Wen, Z.; Li, M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res. 2008, 36, 3025–3030. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-B.; You, Z.-H.; Li, X.; Jiang, T.-H.; Chen, X.; Zhou, X.; Wang, L. Predicting protein–protein interactions from protein sequences by a stacked sparse autoencoder deep neural network. Mol. Biosyst. 2017, 13, 1336–1344. [Google Scholar] [CrossRef]
- Wang, Y.; You, Z.; Xiao, L.; Xing, C.; Jiang, T.; Zhang, J. PCVMZM: Using the Probabilistic Classification Vector Machines Model Combined with a Zernike Moments Descriptor to Predict Protein–Protein Interactions from Protein Sequences. Int. J. Mol. Sci. 2017, 18, 1029. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.B.; You, Z.H.; Li, L.P.; Huang, Y.A.; Yi, H.C. Detection of Interactions between Proteins by Using Legendre Moments Descriptor to Extract Discriminatory Information Embedded in PSSM. Molecules 2017, 22, 1366. [Google Scholar] [CrossRef] [PubMed]
- Ganapathiraju, M.; Balakrishnan, N.; Reddy, R.; Klein-Seetharaman, J. Computational Biology and Language; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Keshava Prasad, T.S.; Goel, R.; Kandasamy, K.; Keerthikumar, S.; Kumar, S.; Mathivanan, S.; Telikicherla, D.; Raju, R.; Shafreen, B.; Venugopal, A. Human Protein Reference Database-2009 update. Nucleic Acids Res. 2009, 37, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Roe, D.; Faulon, J.-L. Predicting protein–protein interactions using signature products. Bioinformatics 2004, 21, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. PICKLE 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T. Subword Regularization: Improving Neural Network Translation Models with Multiple Subword Candidates. arXiv, 2018; arXiv:1804.10959. [Google Scholar]
- Ryan, M.S.; Nudd, G.R. The Viterbi Algorithm. Proc IEEE 1993, 61, 268–278. [Google Scholar]
- Do, C.B.; Batzoglou, S. What is the expectation maximization algorithm? Nat. Biotechnol. 2008, 26, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Sennrich, R.; Haddow, B.; Birch, A. Neural machine translation of rare words with subword units. arXiv, 2015; arXiv:1508.07909. [Google Scholar]
- Abouelhoda, M.I.; Kurtz, S.; Ohlebusch, E. Replacing suffix trees with enhanced suffix arrays. J. Discret. Algorithms 2004, 2, 53–86. [Google Scholar] [CrossRef] [Green Version]
- Kudo, T.; Richardson, J. SentencePiece: A simple and language independent subword tokenizer and detokenizer for Neural Text Processing. arXiv, 2018; arXiv:1808.06226. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient Estimation of Word Representations in Vector Space. arxiv, 2013; arXiv:1301.3781. [Google Scholar]
- Le, Q.V.; Mikolov, T. Distributed Representations of Sentences and Documents. Proc. Mach. Learn. Res. 2014, 32, 1188–1196. [Google Scholar]
- Gittens, A.; Achlioptas, D.; Mahoney, M.W. Skip-Gram − Zipf + Uniform = Vector Additivity. In Proceedings of the 55th Annual Meeting of the Association for Computational Linguistics (Volume 1: Long Papers), Vancouver, BC, Canada, 30 July–4 August 2017; Association for Computational Linguistics, 2017; Volume 1, pp. 69–76. [Google Scholar]
- Wang, P.; Xu, B.; Xu, J.; Tian, G.; Liu, C.; Hao, H. Semantic expansion using word embedding clustering and convolutional neural network for improving short text classification. Neurocomputing 2016, 174, 806–814. [Google Scholar] [CrossRef]
- Rios, A.; Kavuluru, R. Convolutional Neural Networks for Biomedical Text Classification: Application in Indexing Biomedical Articles. In Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics, Atlanta, Georgia, 9–12 September 2015; ACM: New York, NY, USA, 2015; pp. 258–267. [Google Scholar]
- Ren, X.; Zhou, Y.; He, J.; Chen, K.; Yang, X.; Sun, J. A Convolutional Neural Network Based Chinese Text Detection Algorithm via Text Structure Modeling. IEEE Trans. Multimed. 2017, 19, 506–518. [Google Scholar] [CrossRef]
- Du, J.H. Automatic text classification algorithm based on Gauss improved convolutional neural network. J. Comput. Sci. 2017, 21, 195–200. [Google Scholar] [CrossRef]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout: A simple way to prevent neural networks from overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Asgari, E.; Mofrad, M.R. Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics. PLoS ONE 2015, 10, e0141287. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; You, Z.; Li, L.; Cheng, L.; Zhou, X.; Zhang, L.; Li, X.; Jiang, T. Predicting Protein Interactions Using a Deep Learning Method-Stacked Sparse Autoencoder Combined with a Probabilistic Classification Vector Machine. Complexity 2018, 2018, 12. [Google Scholar] [CrossRef]
- Yang, L.; Xia, J.F.; Gui, J. Prediction of protein-protein interactions from protein sequence using local descriptors. Protein Pept. Lett. 2010, 17, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- You, Z.H.; Lei, Y.K.; Zhu, L.; Xia, J.; Wang, B. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinform. 2013, 14, S10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, J.R.; Gough, D.A. Whole-proteome interaction mining. Bioinformatics 2003, 19, 125–135. [Google Scholar] [CrossRef]
- Faulon, J.L.; Faulon, J.L.; Faulon, J.L. Predicting Protein—Protein Interactions Using Signature Products; Oxford University Press: Oxford, UK, 2005. [Google Scholar]
- Nanni, L.; Lumini, A. An ensemble of K-local hyperplanes for predicting protein–protein interactions. Bioinformatics 2006, 22, 1207–1210. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Testing Set | Accu (%) | Sens (%) | Prec (%) | MCC (%) | AUC |
|---|---|---|---|---|---|---|
| Bio2Vec-based | Human | 97.31 | 96.28 | 98.48 | 94.76 | 0.9961 |
| S. cerevisiae | 93.30 | 92.70 | 93.55 | 87.49 | 0.9720 | |
| H. pylori | 88.01 | 89.61 | 87.90 | 78.71 | 0.9394 | |
| Extended-Human | 99.58 | 99.64 | 99.50 | 99.16 | 0.9995 | |
| 3-mers-based | Human | 92.18 | 86.85 | 97.77 | 85.53 | 0.9647 |
| S. cerevisiae | 90.26 | 88.14 | 91.65 | 82.38 | 0.9621 | |
| H. pylori | 83.22 | 89.61 | 80.70 | 82.38 | 0.8924 | |
| Extended-Human | 98.47 | 100 | 96.98 | 96.99 | 0.9998 |
| Model | Accu (%) | Sens (%) | Prec (%) | MCC (%) |
|---|---|---|---|---|
| LDA + RF [32] | 96.40 | 94.20 | N/A | 92.80 |
| LDA + RoF [32] | 95.70 | 97.60 | N/A | 91.80 |
| LDA + SVM [32] | 90.70 | 89.70 | N/A | 81.30 |
| AC + RF [32] | 95.50 | 94.00 | N/A | 91.40 |
| AC + RoF [32] | 95.10 | 93.30 | N/A | 91.10 |
| AC + SVM [32] | 89.30 | 94.00 | N/A | 79.20 |
| Proposed Method | 97.31 | 96.28 | 98.48 | 94.76 |
| Model | Accu (%) | Sens (%) | Prec (%) | MCC (%) |
|---|---|---|---|---|
| ACC [9] | 89.33 | 89.93 | 88.87 | N/A |
| AC [9] | 87.36 | 87.30 | 87.82 | N/A |
| Code1 [33] | 75.08 | 75.81 | 74.75 | N/A |
| Code2 [33] | 80.04 | 76.77 | 82.17 | N/A |
| Code3 [33] | 80.41 | 78.14 | 81.66 | N/A |
| Code4 [33] | 86.15 | 81.03 | 90.24 | N/A |
| PCA-EELM [34] | 87.00 | 86.15 | 87.59 | 77.36 |
| Proposed Method | 93.30 | 92.70 | 93.55 | 87.49 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; You, Z.-H.; Yang, S.; Li, X.; Jiang, T.-H.; Zhou, X. A High Efficient Biological Language Model for Predicting Protein–Protein Interactions. Cells 2019, 8, 122. https://doi.org/10.3390/cells8020122
Wang Y, You Z-H, Yang S, Li X, Jiang T-H, Zhou X. A High Efficient Biological Language Model for Predicting Protein–Protein Interactions. Cells. 2019; 8(2):122. https://doi.org/10.3390/cells8020122
Chicago/Turabian StyleWang, Yanbin, Zhu-Hong You, Shan Yang, Xiao Li, Tong-Hai Jiang, and Xi Zhou. 2019. "A High Efficient Biological Language Model for Predicting Protein–Protein Interactions" Cells 8, no. 2: 122. https://doi.org/10.3390/cells8020122
APA StyleWang, Y., You, Z. -H., Yang, S., Li, X., Jiang, T. -H., & Zhou, X. (2019). A High Efficient Biological Language Model for Predicting Protein–Protein Interactions. Cells, 8(2), 122. https://doi.org/10.3390/cells8020122