Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neutral Lipid Storage Disorders
2.1. Neutral Lipid Storage Disorder with Myopathy
2.2. Neutral Lipid Storage Disorder with Ichthyosis
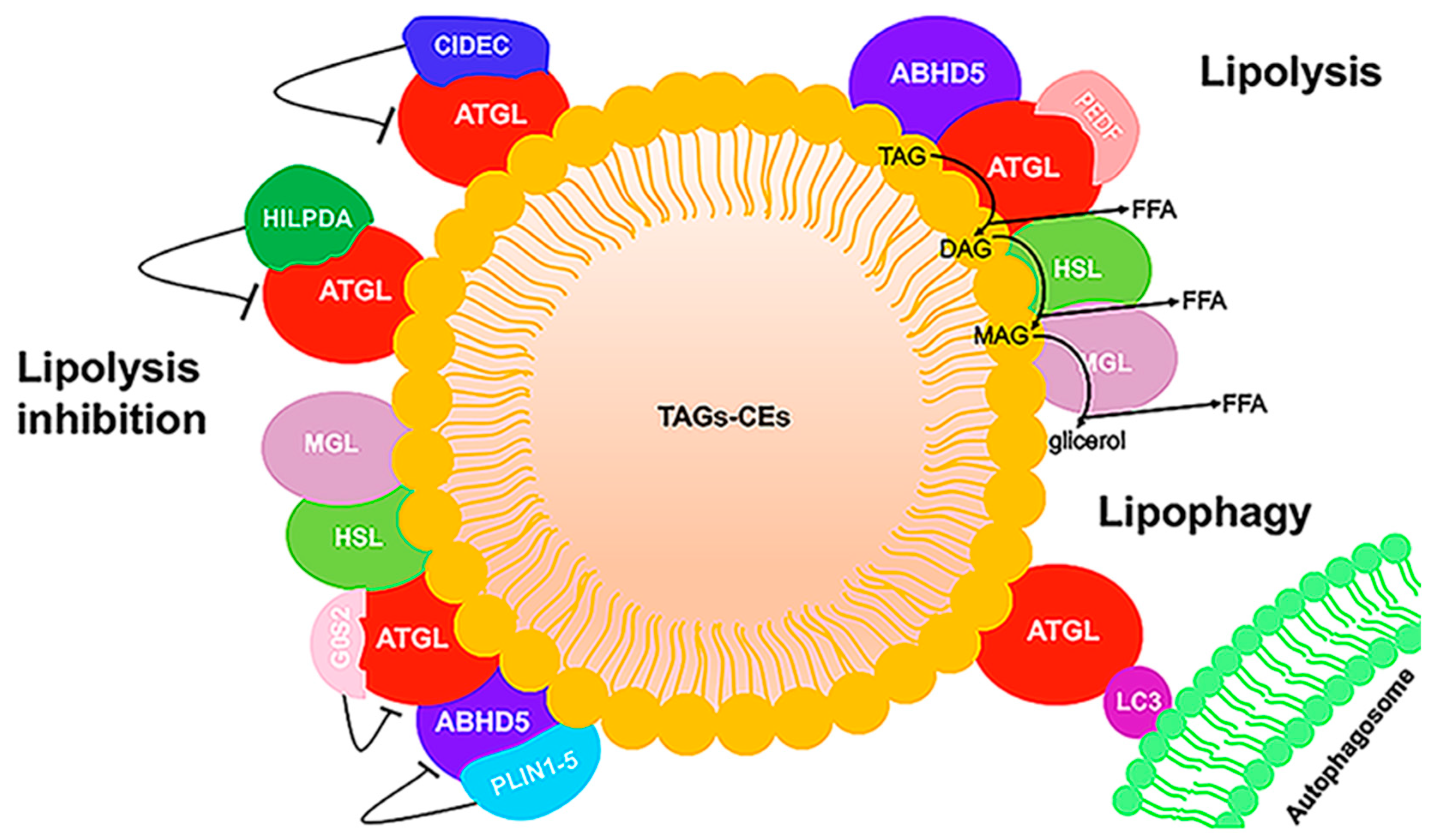
2.3. ATGL and ABHD5 Proteins in LD
3. LDs in NLSD Patient Tissues
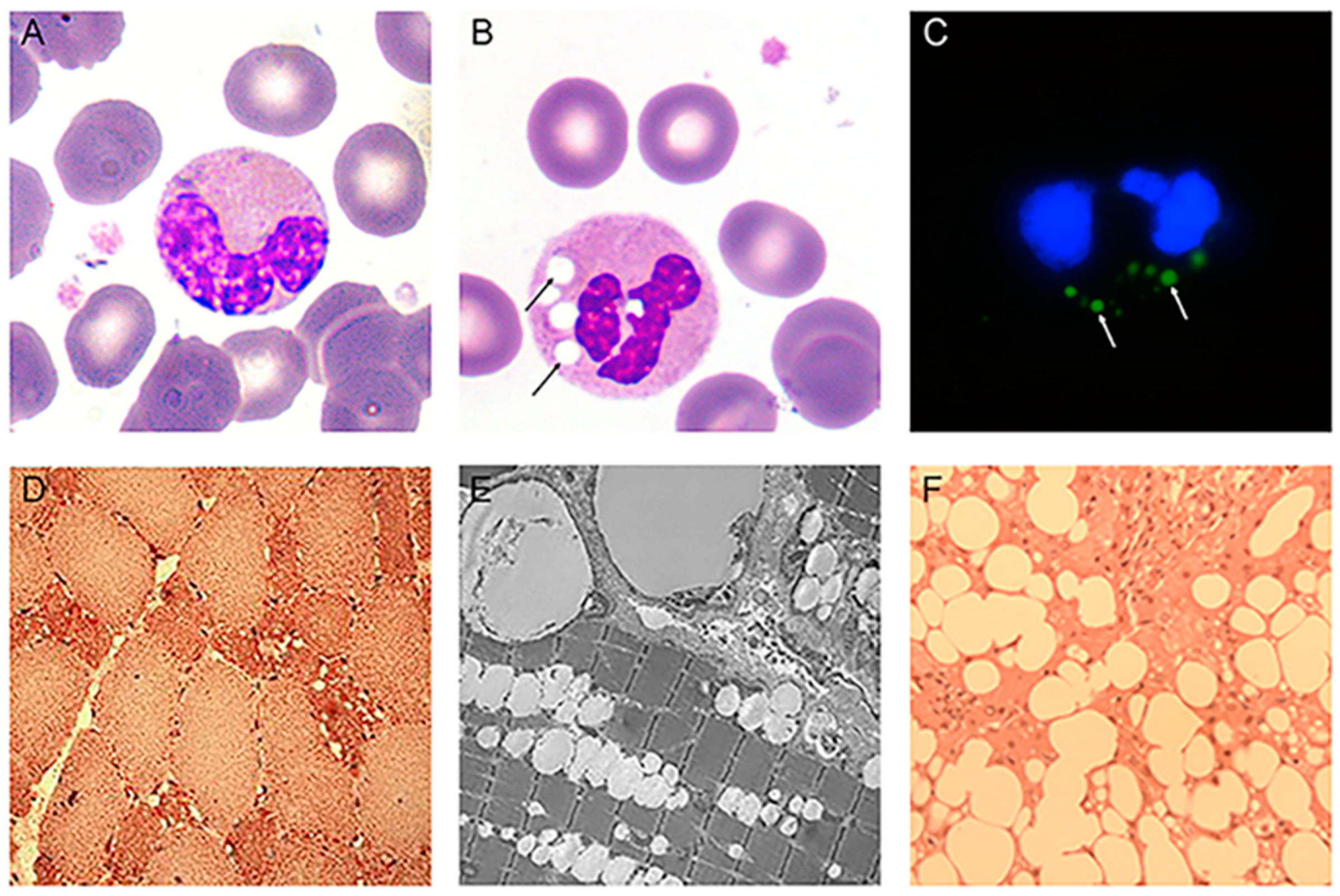
3.1. Jordans’ Anomaly
3.2. Muscle
3.3. Heart
3.4. Liver
3.5. Skin
4. NLSD Cellular Models
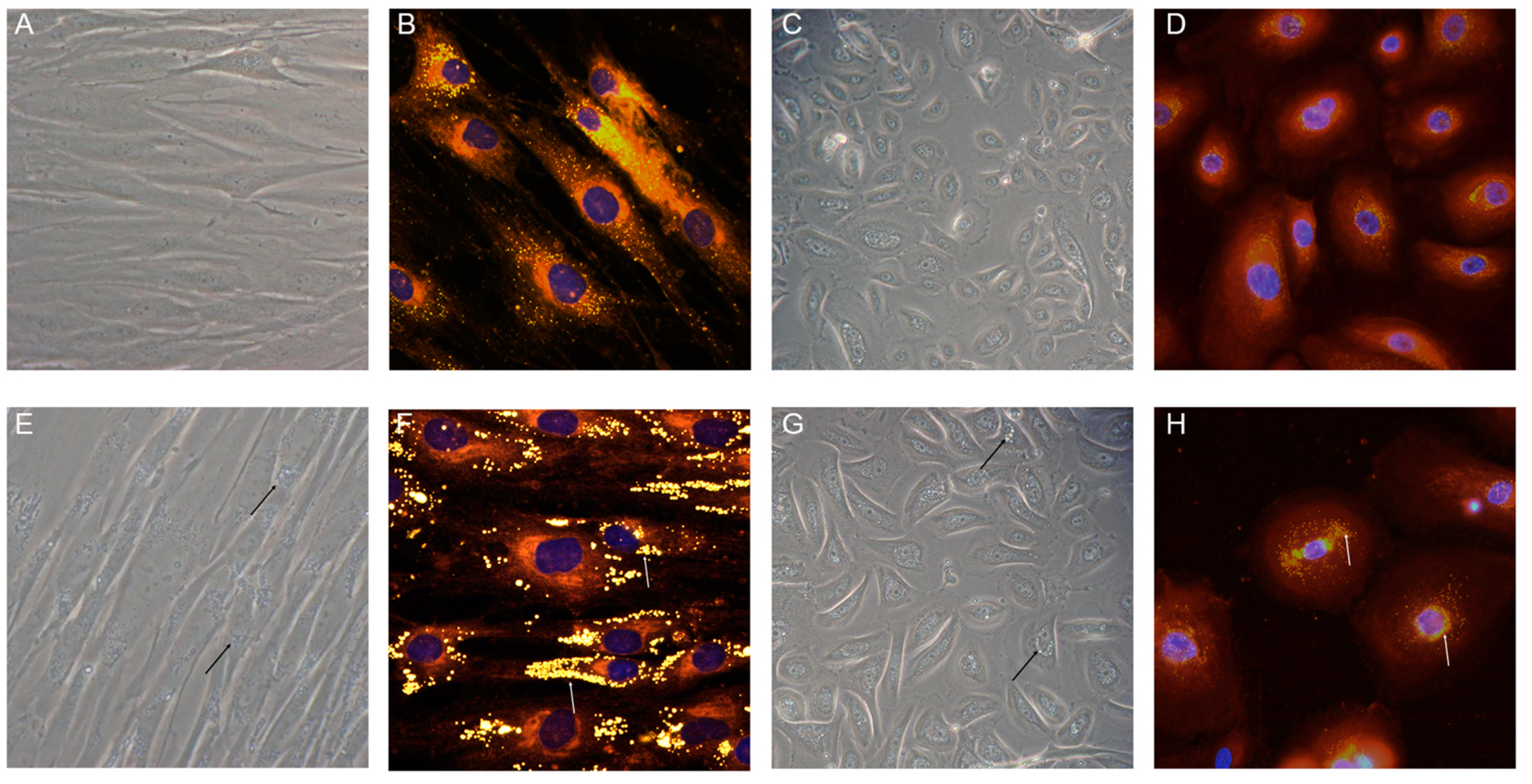
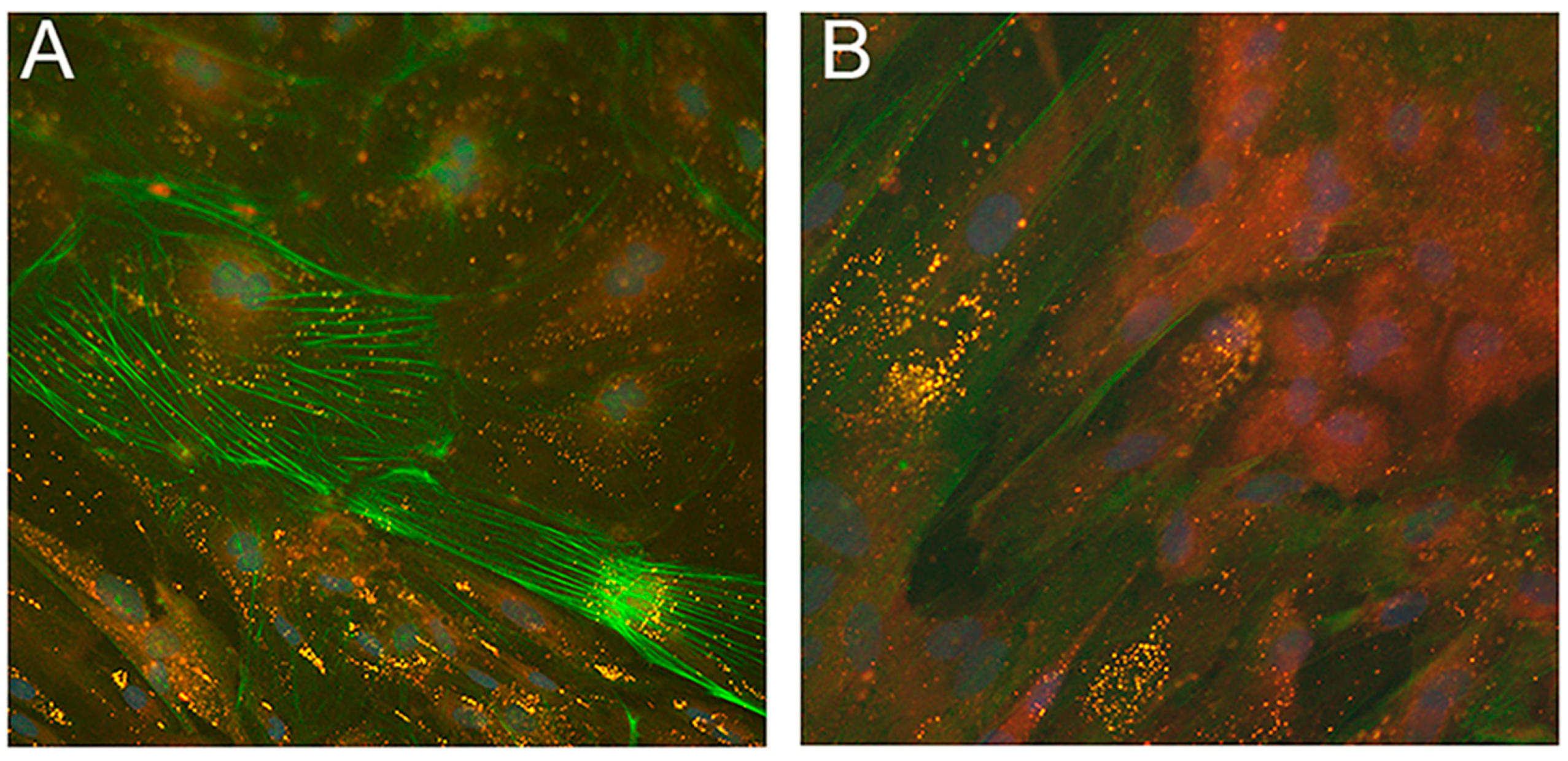
4.1. Historical Experiments Performed on Primary Dermal NLSD Fibroblasts
4.2. Studies of Cultured Fibroblasts
4.3. Induced Pluripotent Stem Cells
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Walther, T.C.; Farese, R.V., Jr. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R.; Zimmermann, R.; Eichmann, T.O.; Kohlwein, S.D.; Haemmerle, G.; Lass, A.; Madeo, F. FAT SIGNALS—Lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.J. The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma 2012, 249, 541–585. [Google Scholar] [CrossRef]
- Thiam, A.R.; Farese, R.V., Jr.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerner, J.; Hoppel, C. Fatty acid import into mitochondria. Biochim. Biophys. Acta 2000, 1486, 1–17. [Google Scholar] [CrossRef]
- Eaton, S. Control of mitochondrial beta-oxidation flux. Prog. Lipid. Res. 2002, 41, 197–239. [Google Scholar] [CrossRef]
- Mazzali, G.; Fantin, F.; Zoico, E.; Sepe, A.; Bambace, C.; Faccioli, S.; Pedrotti, M.; Corzato, F.; Rizzatti, V.; Faggian, G.; et al. Heart fat infiltration in subjects with and without coronary artery disease. J. Clin. Endocrinol. Metab. 2015, 100, 3364–3371. [Google Scholar] [CrossRef]
- Stary, H.C. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am. J. Clin. Nutr. 2000, 72, 1297S–1306S. [Google Scholar] [CrossRef]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef]
- Fischer, J.; Lefèvre, C.; Morava, E.; Mussini, J.M.; Laforêt, P.; Negre-Salvayre, A.; Lathrop, M.; Salvayre, R. The gene encoding adipose triglyceride lipase (PNPLA2) is mutated in neutral lipid storage disease with myopathy. Nat. Genet. 2007, 39, 28–30. [Google Scholar] [CrossRef]
- Lefevre, C.; Jobard, F.; Caux, F.; Bouadjar, B.; Karaduman, A.; Heilig, R.; Lakhdar, H.; Wollenberg, A.; Verret, J.L.; Weissenbach, J.; et al. Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am. J. Hum. Genet. 2001, 69, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, R470–R481. [Google Scholar] [CrossRef] [PubMed]
- Welte, M.A.; Gould, A.P. Lipid droplet functions beyond energy storage. Biochim. Biophys. Acta 2017, 1862, 1260–1272. [Google Scholar] [CrossRef] [PubMed]
- Grumet, L.; Taschler, U.; Lass, A. Hepatic Retinyl Ester Hydrolases and the Mobilization of Retinyl Ester Stores. Nutrients 2016, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Kayden, H.J. Tocopherol distribution and intracellular localization in human adipose tissue. Am. J. Clin. Nutr. 1987, 46, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Fujikawa, N.; Nimura, S.; Tokuoka, Y.; Tsuda, S.; Aiuchi, T.; Kato, R.; Obama, T.; Itabe, H. Characterization of lipid droplets in steroidogenic MLTC-1 Leydig cells: Protein profiles and the morphological change induced by hormone stimulation. Biochim. Biophys. Acta 2015, 1851, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Dichlberger, A.; Schlager, S.; Maaninka, K.; Schneider, W.J.; Kovanen, P.T. Adipose triglyceride lipase regulates eicosanoid production in activated human mast cells. J. Lipid Res. 2014, 55, 2471–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartz, R.; Li, W.H.; Venables, B.; Zehmer, J.K.; Roth, M.R.; Welti, R.; Anderson, R.G.; Liu, P.; Chapman, K.D. Lipidomics reveals that adiposomes store ether lipids and mediate phospholipid traffic. J. Lipid Res. 2007, 48, 837–847. [Google Scholar] [CrossRef]
- Murphy, G., Jr.; Rouse, R.L.; Polk, W.W.; Henk, W.G.; Barker, S.A.; Boudreaux, M.J.; Floyd, Z.E.; Penn, A.L. Combustion-derived hydrocarbons localize to lipid droplets in respiratory cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 532–540. [Google Scholar] [CrossRef]
- Bourez, S.; Le Lay, S.; Van den Daelen, C.; Louis, C.; Larondelle, Y.; Thome, J.P.; Schneider, Y.J.; Dugail, I.; Debier, C. Accumulation of polychlorinated biphenyls in adipocytes: Selective targeting to lipid droplets and role of caveolin-1. PLoS ONE 2012, 7, e31834. [Google Scholar] [CrossRef]
- Bosma, M.; Dapito, D.H.; Drosatos-Tampakaki, Z.; Huiping-Son, N.; Huang, L.S.; Kersten, S.; Drosatos, K.; Goldberg, I.J. Sequestration of fatty acids in triglycerides prevents endoplasmic reticulum stress in an in vitro model of cardiomyocyte lipotoxicity. Biochim. Biophys. Acta 2014, 1841, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; Simon, M.C. HIF2alpha-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Camus, G.; Herker, E.; Modi, A.A.; Haas, J.T.; Ramage, H.R.; Farese, R.V., Jr.; Ott, M. Diacylglycerol acyltransferase-1 localizes hepatitis C virus NS5A protein to lipid droplets and enhances NS5A interaction with the viral capsid core. J. Biol. Chem. 2013, 288, 9915–9923. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, N.G.; Mondotte, J.A.; Byk, L.A.; De Maio, F.A.; Samsa, M.M.; Alvarez, C.; Gamarnik, A.V. Dengue Virus Uses a Non-Canonical Function of the Host GBF1-Arf-COPI System for Capsid Protein Accumulation on Lipid Droplets. Traffic 2015, 16, 962–977. [Google Scholar] [CrossRef]
- Gaunt, E.R.; Zhang, Q.; Cheung, W.; Wakelam, M.J.; Lever, A.M.; Desselberger, U. Lipidome analysis of rotavirus-infected cells confirms the close interaction of lipid droplets with viroplasms. J. Gen. Virol. 2013, 94, 1576–1586. [Google Scholar] [CrossRef] [Green Version]
- Gallardo-Montejano, V.I.; Saxena, G.; Kusminski, C.M.; Yang, C.; McAfee, J.L.; Hahner, L.; Hoch, K.; Dubinsky, W.; Narkar, V.A.; Bickel, P.E. Nuclear Perilipin 5 integrates lipid droplet lipolysis with PGC-1alpha/SIRT1-dependent transcriptional regulation of mitochondrial function. Nat. Commun. 2016, 7, 12723. [Google Scholar] [CrossRef]
- Aramburu, J.; Drews-Elger, K.; Estrada-Gelonch, A.; Minguillon, J.; Morancho, B.; Santiago, V.; Lopez-Rodriguez, C. Regulation of the hypertonic stress response and other cellular functions by the Rel-like transcription factor NFAT5. Biochem. Pharmacol. 2006, 72, 1597–1604. [Google Scholar] [CrossRef]
- Fujimoto, T.; Ohsaki, Y. Proteasomal and autophagic pathways converge on lipid droplets. Autophagy 2006, 2, 299–301. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Walther, T.C. Lipid droplets go nuclear. J. Cell Biol. 2016, 212, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordans, C.H. The familial occurrence of fat containing vacuoles in the leukocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva. Acta Med. Scand. 1953, 145, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Rozenszajn, L.; Klajman, A.; Yaffe, D.; Efiiati, P. Jordans’ anomaly in white blood cells. Report of case. Blood 1966, 28, 258–265. [Google Scholar] [PubMed]
- Igal, R.A.; Rhoads, J.M.; Coleman, R.A. Neutral Lipid Storage Disease with Fatty Liver and Cholestasis. J. Pediatr. Gastroenterol. Nutr. 1997, 25, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Reilich, P.; Horvath, R.; Krause, S.; Schramm, N.; Turnbull, D.M.; Trenell, M.; Hollingsworth, K.G.; Gorman, G.S.; Hans, V.H.; Reimann, J.; et al. The phenotypic spectrum of neutral lipid storage myopathy due to mutations in the PNPLA2 gene. J. Neurol. 2011, 258, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Laforêt, P.; Stojkovic, T.; Bassez, G.; Carlier, P.G.; Clément, K.; Wahbi, K.; Petit, F.M.; Eymard, B.; Carlier, R.Y. Neutral lipid storage disease with myopathy: A whole-body nuclear MRI and metabolic study. Mol. Genet. Metab. 2013, 108, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Kuroda, H.; Izumi, R.; Tateyama, M.; Kato, M.; Sugimura, K.; Sakata, Y.; Ikeda, Y.; Hirano, K.; Aoki, M. A novel mutation in PNPLA2 causes neutral lipid storage disease with myopathy and triglyceride deposit cardiomyovasculopathy: A case report and literature review. Neuromuscul. Disord. 2014, 24, 634–641. [Google Scholar] [CrossRef]
- Pennisi, E.M.; Arca, M.; Bertini, E.S.; Bruno, C.; Cassandrini, D.; D’amico, A.; Garibaldi, M.; Gragnani, F.; Maggi, L.; Massa, R.; et al. Neutral Lipid Storage Diseases: clinical/genetic features and natural history in a large cohort of Italian patients. Orphanet. J. Rare Dis. 2017, 90, 1–10. [Google Scholar] [CrossRef]
- Akiyama, M.; Sakai, K.; Ogawa, M.; McMillan, J.R.; Sawamura, D.; Shimizu, H. Novel duplication mutation in the patatin domain of adipose triglyceride lipase (PNPLA2) in neutral lipid storage disease with severe myopathy. Muscle Nerve 2007, 36, 856–859. [Google Scholar] [CrossRef]
- Campagna, F.; Nanni, L.; Quagliarini, F.; Pennisi, E.; Michailidis, C.; Pierelli, F.; Bruno, C.; Casali, C.; DiMauro, S.; Arca, M. Novel mutations in the adipose triglyceride lipase gene causing neutral lipid storage disease with myopathy. Biochem. Biophys. Res. Commun. 2008, 377, 843–846. [Google Scholar] [CrossRef]
- Hirano, K.; Ikeda, Y.; Zaima, N.; Sakta, Y.; Matsumiya, G. Triglyceride deposit cardiomyovasculopathy. N. Engl. J. Med. 2008, 359, 2396–2398. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Inoguchi, T.; Maeda, Y.; Nakashima, N.; Kuwano, A.; Eto, E.; Ueno, N.; Sasaki, S.; Sawada, F.; Fujii, M.; et al. The lack of the C-terminal domain of adipose triglyceride lipase causes neutral lipid storage disease through impaired interactions with lipid droplets. J. Clin. Endocrinol. Metab. 2008, 93, 2877–2884. [Google Scholar] [CrossRef] [PubMed]
- Ohkuma, A.; Nonaka, I.; Malicdan, M.C.; Noguchi, S.; Ohji, S.; Nomura, K.; Sugie, H.; Hayashi, Y.K.; Nishino, I. Distal lipid storage myopathy due to PNPLA2 mutation. Neuromuscul. Disord. 2008, 18, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hong, D.; Wang, Z.; Yuan, Y. A novel PNPLA2 mutation causes neutral lipid storage disease with myopathy (NLSDM) presenting muscular dystrophic features with lipid storage and rimmed vacuoles. Clin. Neuropathol. 2010, 29, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Davidzon, G.; Tanji, K.; Macdermott, E.J.; Larsen, L.; Davidson, M.M.; Haller, R.G.; Szczepaniak, L.S.; Lehman, T.J.; Hirano, M.; et al. Neutral lipid storage disease with subclinical myopathy due to a retrotransposal insertion in the PNPLA2 gene. Neuromuscul. Disord. 2010, 20, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Ash, D.B.; Papadimitriou, D.; Hays, A.P.; Dimauro, S.; Hirano, M. A novel mutation in PNPLA2 leading to neutral lipid storage disease with myopathy. Arch. Neurol. 2012, 69, 1190–1192. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Li, W.; Wen, B.; Zhao, Y.; Fenster, D.S.; Wang, Y.; Gong, Y.; Yan, C. Novel PNPLA2 gene mutations in Chinese Han patients causing neutral lipid storage disease with myopathy. J. Hum. Genet. 2012, 57, 679–681. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, C.; Brisca, G.; Cassandrini, D.; Scapolan, S.; Astrea, G.; Valle, M.; Scuderi, F.; Trucco, F.; Natali, A.; Magnano, G.; et al. Subclinical myopathy in a child with neutral lipid storage disease and mutations in the PNPLA2 gene. Biochem. Biophys. Res. Commun. 2013, 430, 241–244. [Google Scholar] [CrossRef]
- Hirano, K.; Tanaka, T.; Ikeda, Y.; Yamaguchi, S.; Zaima, N.; Kobayashi, K.; Suzuki, A.; Sakata, Y.; Sakata, Y.; Kobayashi, K.; et al. Genetic Mutations in the Adipose Triglyceride Lipase and Myocardial Overexpression of Peroxisome Proliferated Activated Receptor- in Patients with Triglyceride Deposit Cardiomyovasculopathy. Biochem. Biophys. Res. Commun. 2014, 443, 574–579. [Google Scholar] [CrossRef]
- Massa, R.; Pozzessere, S.; Rastelli, E.; Serra, L.; Terracciano, C.; Gibellini, M.; Bozzali, M.; Arca, M. Neutral lipid-storage disease with myopathy and extended phenotype with novel PNPLA2 mutation. Muscle Nerve 2016, 53, 644–648. [Google Scholar] [CrossRef]
- Missaglia, S.; Maggi, L.; Mora, M.; Gibertini, S.; Blasevich, F.; Agostoni, P.; Moro, L.; Cassandrini, D.; Santorelli, F.M.; Gerevini, S.; et al. Late onset of neutral lipid storage disease due to novel PNPLA2 mutations causing a total loss of lipase activity in a patient with myopathy and slight cardiac involvement. Neuromuscul. Disord. 2017, 27, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Pasanisi, M.B.; Missaglia, S.; Cassandrini, D.; Salerno, F.; Farina, S.; Andreini, D.; Agostoni, P.; Morandi, L.; Mora, M.; Tavian, D. Severe cardiomyopathy in a young patient with complete deficiency of adipose triglyceride lipase due to a novel mutation in PNPLA2 gene. Int. J. Cardiol. 2016, 207, 165–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweiger, M.; Lass, A.; Zimmermann, R.; Eichmann, T.O.; Zechner, R. Neutral lipid storage disease: Genetic disorders caused by mutations in adipose triglyceride lipase/PNPLA2 or CGI-58/ABHD5. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Coassin, S.; Schweiger, M.; Kloss-Brandstätter, A.; Lamina, C.; Haun, M.; Erhart, G.; Paulweber, B.; Rahman, Y.; Olpin, S.; Wolinski, H.; et al. Investigation and functional characterization of rare genetic variants in the adipose triglyceride lipase in a large healthy working population. PLoS Genet. 2010, 6, e1001239. [Google Scholar] [CrossRef] [PubMed]
- Tavian, D.; Missaglia, S.; Redaelli, C.; Pennisi, E.M.; Invernici, G.; Wessalowski, R.; Maiwald, R.; Arca, M.; Coleman, R.A. Contribution of novel ATGL missense mutations to the clinical phenotype of NLSD-M: A strikingly low amount of lipase activity may preserve cardiac function. Hum. Mol. Genet. 2012, 21, 5318–5328. [Google Scholar] [CrossRef] [PubMed]
- Missaglia, S.; Tasca, E.; Angelini, C.; Moro, L.; Tavian, D. Novel missense mutations in PNPLA2 causing late onset and clinical heterogeneity of neutral lipid storage disease with myopathy in three siblings. Mol. Genet. Metab. 2015, 115, 110–117. [Google Scholar] [CrossRef]
- Pennisi, E.M.; Missaglia, S.; Di Mauro, S.; Bernardi, C.; Akman, H.O.; Tavia, D. A myopathy with unusual features caused by PNPLA2 gene mutations. Muscle Nerve 2015, 51, 609–613. [Google Scholar] [CrossRef]
- Chanarin, I.; Patel, A.; Slavin, G.; Wills, E.J.; Andrews, T.M.; Stewart, G. Neutral-lipid storage disease: A new disorder of lipid metabolism. Br. Med. J. 1975, 1, 553–555. [Google Scholar] [CrossRef]
- Dorfman, M.L.; Hershko, C.; Eisenberg, S. Ichthyosiform dermatosis with systematic lipidosis. Arch. Dermatol. 1974, 110, 261–266. [Google Scholar] [CrossRef]
- Redaelli, C.; Coleman, A.R.; Moro, L.; Dacou-Voutetakis, C.; Elsayed, S.M.; Prati, D.; Colli, A.; Mela, D.; Colombo, R.; Tavian, D. Clinical and genetic characterization of Chanarin-Dorfman Syndrome patients: first report of large deletions in the ABHD5. Orphanet. J. Rare Dis. 2010, 5, 33. [Google Scholar] [CrossRef]
- Ronchetti, A.; Prati, D.; Pezzotta, M.G.; Tavian, D.; Colombo, R.; Callea, F.; Colli, A. Severe steatohepatitis in a patient with a rare disorder of neutral lipid storage due to a ABDH5 mutation. J. Hepatol. 2008, 49, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Missaglia, S.; Valadares, E.R.; Moro, L.; Faguntes, E.D.; Quintão Roque, R.; Giardina, B.; Tavian, D. Early onset of chanarin-dorfman syndrome with severe liver involvement in a patient with a complex rearrangement of ABHD5 promoter. BMC Med. Genet. 2014, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Gothwal, S.; Satpathy, A.K.; Missaglia, S.; Tavian, D.; Das, P.; Timila, D.; Kabra, M. Chanarin Dorfman Syndrome: A Case Report with Novel Nonsense Mutation. Gene 2016, 575, 359–362. [Google Scholar] [CrossRef]
- Durdu, M.; Missaglia, S.; Moro, L.; Tavian, D. Clinical and genetic characterization of a Chanarin Dorfman Syndrome patient born to diseased parents. BMC Med. Genet. 2018, 19, 88. [Google Scholar] [CrossRef] [PubMed]
- Lass, A.; Zimmermann, R.; Haemmerle, G.; Riederer, M.; Schoiswohl, G.; Schweiger, M.; Kienesberger, P.; Strauss, J.G.; Gorkiewicz, G.; Zechner, R. Adipose triglyceride lipase-mediated lipolysis of cellular fat stores is activated by CGI-58 and defective in Chanarin-Dorfman Syndrome. Cell Metab. 2006, 3, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nur, B.G.; Gencpinar, P.; Yuzbasioglu, A.; Emre, S.D.; Mihci, E. Chanarin Dorfman syndrome: genotype-phenotype correlation. Eur. J. Med. Genet. 2015, 58, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 1941. [Google Scholar] [CrossRef]
- Bersuker, K.; Peterson, C.W.H.; To, M.; Sahl, S.J.; Savikhin, V.; Grossman, E.A.; Nomura, D.K.; Olzmann, J.A. A Proximity Labeling Strategy Provides Insights into the Composition and Dynamics of LipidDroplet Proteomes. Dev. Cell 2018, 44, 97–112. [Google Scholar] [CrossRef]
- Kory, N.; Farese, R.V., Jr.; Walther, T.C. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Wang, Y.; Cui, L.; Deng, Y.; Xu, S.; Yu, J.; Cichello, S.; Serrero, G.; Ying, Y.; Liu, P. Morphologically and Functionally Distinct Lipid Droplet Subpopulations. Sci. Rep. 2016, 6, 29539. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, A.S.; Coleman, R.A.; Kraemer, F.B.; McManaman, J.L.; Obin, M.S.; Puri, V.; Yan, Q.W.; Miyoshi, H.; Mashek, D.G. The role of lipid droplets in metabolic disease in rodents and humans. J. Clin. Investig. 2011, 121, 2102–2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Lu, X.; Lombès, M.; Rha, G.B.; Chi, Y.I.; Guerin, T.M.; Smart, E.J.; Liu, J. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, H.; Perfield, J.W., 2nd; Obin, M.S.; Greenberg, A.S. Adipose triglyceride lipase regulates basal lipolysis and lipid droplet size in adipocytes. J. Cell. Biochem. 2008, 105, 1430–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itabe, H.; Yamaguchi, T.; Nimura, S.; Sasabe, N. Perilipins: A diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Sztalryd, C.; Brasaemle, D.L. The perilipin family of lipid droplet proteins: Gatekeepers of intracellular lipolysis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 Pt B, 1221–1232. [Google Scholar] [CrossRef]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Goodman, J.M. The lipid droplet—A well-connected organelle. Front. Cell Dev. Biol. 2015, 3, 49. [Google Scholar] [CrossRef]
- Cornaciu, I.; Boeszoermenyi, A.; Lindermuth, H.; Nagy, H.M.; Cerk, I.K.; Ebner, C.; Salzburger, B.; Gruber, A.; Schweiger, M.; Zechner, R.; et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS ONE 2011, 6, e26349. [Google Scholar] [CrossRef]
- Zhang, X.; Saarinen, A.M.; Hitosugi, T.; Wang, Z.; Wang, L.; Ho, T.H.; Liu, J. Inhibition of intracellular lipolysis promotes human cancer cell adaptation to hypoxia. eLife 2017, 6, pii:e31132. [Google Scholar] [CrossRef]
- Huang, K.T.; Lin, C.C.; Tsai, M.C.; Chen, K.D.; Chiu, K.W. Pigment epithelium-derived factor in lipid metabolic disorders. Biomed. J. 2018, 41, 102–108. [Google Scholar] [CrossRef]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862 (10 Pt B), 1178–1187. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, S.; Agarwal, S.; Nangia, A.; Chander, R.; Varghese, B. Neutral lipid storage disease with unusual presentation: Report of three cases. Pediatr. Dermatol. 2012, 29, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Moyano, E.; Godoy-Diaz, D.J.; Ponce-Verdugo, L.; Sanz-Trelles, A.; Vera-Casaño, A.; Sierra-Salinas, C. Chanarin–Dorfman syndrome in three siblings in a non-consanguineous family. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 157–159. [Google Scholar] [CrossRef] [PubMed]
- Mela, D.; Artom, A.; Goretti, R.; Varagona, G.; Riolfo, M.; Ardoino, S.; Sanguineti, G.; Vitali, A.; Ricciardi, S. Dorfman-Chanarin syndrome: A case with prevalent hepatic involvement. J. Hepatol. 1996, 25, 769–771. [Google Scholar] [CrossRef]
- Tavian, D.; Colombo, R. Improved cytochemical method for detecting Jordans’ bodies in neutral-lipid storage diseases. J. Clin. Pathol. 2007, 60, 956–958. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Nagasaka, H.; Ochi, Y.; Kobayashi, K.; Nakamura, H.; Nakatani, D.; Yamaguchi, S.; Yamaki, S.; Wada, A.; Shirata, Y.; et al. Peripheral leukocyte anomaly detected with routine automated hematology analyzer sensitive to adipose triglyceride lipase deficiency manifesting neutral lipid storage disease with myopathy/triglyceride deposit cardiomyovasculopathy. Mol. Genet. Metab. Rep. 2014, 1, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Bosma, M. Lipid droplet dynamics in skeletal muscle. Exp. Cell Res. 2016, 340, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M.; Schuldiner, M. Stepping outside the comfort zone of membrane contact site research. Nat. Rev. Mol. Cell Biol. 2018, 19, 483–484. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.E.; Bucarey, J.L.; Espinosa, A. Muscle Lipid Metabolism: Role of Lipid Droplets and Perilipins. J. Diabetes Res. 2017, 2017, 1789395. [Google Scholar] [CrossRef]
- Higashi, M.; Hirano, K.; Kobayashi, K.; Ikeda, Y.; Issiki, A.; Otsuka, T.; Suzuki, A.; Yamaguchi, S.; Zaima, N.; Hamada, S.; et al. Distinct cardiac phenotype between two homozygotes born in a village with accumulation of a genetic deficiency of adipose triglyceride lipase. Int. J. Cardiol. 2015, 192, 30–32. [Google Scholar] [CrossRef]
- Schreiber, R.; Xie, X.; Schweiger, M. Of mice and men: The physiological role of adipose triglyceride lipase (ATGL). Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, pii: S1388-1981(18)30335-4. [Google Scholar] [CrossRef] [PubMed]
- Gluchowsky, N.L.; Becuwe, M.; Walther, T.C.; Farese, R.V., Jr. Lipid droplets and liver disease: From basic biology to clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Aggarwal, P.; Dhawan, J.; Singh, U.R.; Bhattacharya, S.N. Dorfman-Chanarin syndrome. Indian J. Dermatol. Venereol. Leprol. 2007, 73, 36–39. [Google Scholar] [CrossRef] [PubMed]
- Pujol, R.M.; Gilaberte, M.; Toll, A.; Florensa, L.; Lloreta, J.; González-Enseñat, M.A.; Fischer, J.; Azon, A. Erythrokeratoderma variabilis-like ichthyosis in Chanarin–Dorfman syndrome. Br. J. Dermatol. 2005, 153, 838–841. [Google Scholar] [CrossRef] [PubMed]
- Srebrnik, A.; Brenner, S.; Ilie, B.; Messer, G. Dorfman–Chanarin syndrome: morphologic studies and presentation of new cases. Am. J. Dermatopathol. 1998, 20, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Demir, B.; Sen, A.; Bilik, L.; Deveci, U.; Ozercan, I.H.; Cicek, D.; Dogan, Y. Chanarin-Dorfman syndrome. Clin. Exp. Dermatol. 2017, 42, 699–701. [Google Scholar] [CrossRef] [PubMed]
- Aksu, G.; Kalkan Ucar, S.; Bulut, Y.; Aydinok, Y.; Sen, S.; Anal, O.; Simsek Gosen, D.; Darcan, S.; Coker, M.; Kutukculer, N. Renal involvement as a rare complication of Dorfman-Chanarin syndrome: A case report. Pediatr. Dermatol. 2008, 25, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Nara, A.; Nakamichi, S.; Kihara, A. Molecular mechanism of the ichthyosis pathology of Chanarin-Dorfman syndrome: Stimulation of PNPLA1-catalyzed ω-O-acylceramide production by ABHD5. J. Dermatol. Sci. 2018, 92, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Kien, B.; Grond, S.; Haemmerle, G.; Lass, A.; Eichmann, T.O.; Radner, F.P.W. ABHD5 stimulates PNPLA1-mediated ω-O-acylceramide biosynthesis essential for a functional skin permeability barrier. J. Lipid Res. 2018, 59, 2360–2367. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Sawamura, D.; Nomura, Y.; Sugawara, M.; Shimizu, H. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin syndrome. J. Investig. Dermatol. 2003, 121, 1029–1034. [Google Scholar] [CrossRef]
- Akiyama, M.; Sakai, K.; Takayama, C.; Yanagi, T.; Yamanaka, Y.; McMillan, J.R.; Shimizu, H. CGI-58 is an alpha/beta-hydrolase within lipid transporting lamellar granules of differentiated keratinocytes. Am. J. Pathol. 2008, 173, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Cho, Y.; Moradian, S.; Kim, J.; Nakajima, K.; Crumrine, D.; Park, K.; Ujihara, M.; Akiyama, M.; Shimizu, H.; et al. Neutral lipid storage leads to acylceramide deficiency, likely contributing to the pathogenesis of Dorfman-Chanarin syndrome. J. Investig. Dermatol. 2010, 130, 2497–2499. [Google Scholar] [CrossRef]
- Miranda, A.; DiMauro, S.; Eastwood, A.; Hays, A.; Johnson, W.G.; Olarte, M.; WhitIock, R.; Mayeux, R.; Rowland, L.P. Lipid storage, myopathy, ichthyosis and steatorrhea. Muscle Nerve 1979, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C. Multisystem triglyceride storage disorder with impaired long-chain fatty acid oxidation. Ann. Neurol. 1980, 7, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.L.; Monger, D.J.; Rutherford, S.L.; Hincenbergs, M.; Rehfeld, S.J.; Grunfeld, C. Neutral lipid storage disease with ichthyosis: Lipid content and metabolism of fibroblasts. J. Inherit. Metab. Dis. 1988, 11, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Radom, J.; Salvayre, R.; Mussini, J.M.; De Lisle, B.; Negre, A.; Maret, A.; Billaudel, S.; Douste-Blazy, L. Biochemical and ultrastructural features of human fibroblasts cultured from a new variant of type 3 lipid storage myopathy. Biol. Cell 1988, 62, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Hilaire, N.; Nègre-Salvayre, A.; Salvayre, R. Cytoplasmic triacylglycerols and cholesteryl esters are degraded in two separate catabolic pools in cultured human fibroblasts. FEBS Lett. 1993, 328, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Hilaire, N.; Nègre-Salvayre, A.; Salvayre, R. Cellular uptake and catabolism of high-density-lipoprotein triacylglycerols in human cultured fibroblasts: degradation block in neutral lipid storage disease. Biochem. J. 1994, 297, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Igal, R.A.; Coleman, R.A. Acylglycerol recycling from triacylglycerol to phospholipid, not lipase activity, is defective in Neutral Lipid Storage Disease fibroblasts. J. Biol. Chem. 1996, 271, 16644–16651. [Google Scholar] [CrossRef]
- Igal, R.A.; Coleman, R.A. Neutral lipid storage disease: A genetic disorder with abnormalities in the regulation of phospholipid metabolism. J. Lipid Res. 1998, 39, 31–43. [Google Scholar]
- Zimmermann, R.; Strauss, J.G.; Haemmerle, G.; Schoiswohl, G.; Birner-Gruenberger, R.; Riederer, M.; Lass, A.; Neuberger, G.; Eisenhaber, F.; Hermetter, A.; et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004, 306, 1383–1386. [Google Scholar] [CrossRef] [PubMed]
- Villena, J.A.; Roy, S.; Sarkadi-Nagy, E.; Kim, K.H.; Sul, H.S. Desnutrin, an adipocyte gene encoding a novel patatin domaincontaining protein, is induced by fasting and glucocorticoids: Ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 2004, 279, 47066–47075. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef] [PubMed]
- Langin, D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol. Res. 2006, 53, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.A.; Ben Ali, Y.; Abdelkafi, S.; Mendoza, L.D.; Leclaire, J.; Fotiadu, F.; Buono, G.; Carrière, F.; Abousalham, A. In vitro stereoselective hydrolysis of diacylglycerols by hormone-sensitive lipase. Biochim. Biophys. Acta 2010, 1801, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.P.; Chuang, C.Y.; Kuo, H.C. Induced pluripotent stem cell technology for disease modeling and drug screening with emphasis on lysosomal storage diseases. Stem Cell Res. Ther. 2011, 3, 34. [Google Scholar] [CrossRef] [PubMed]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling neuronal defects associated with a lysosomal disorder using patient-derived induced pluripotent stem cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef] [Green Version]
- Tiscornia, G.; Vivas, E.L.; Matalonga, L. Neuronopathic Gaucher’s disease: Induced pluripotent stem cells for disease modelling and testing chaperone activity of small compounds. Hum. Mol. Genet. 2013, 22, 633–645. [Google Scholar] [CrossRef]
- Dimos, J.T.; Rodolfa, K.T.; Niakan, K.K.; Weisenthal, L.M.; Mitsumoto, H.; Chung, W.; Croft, G.F.; Saphier, G.; Leibel, R.; Goland, R.; et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 2008, 321, 1218–1221. [Google Scholar] [CrossRef]
- Wernig, M.; Zhao, J.P.; Pruszak, J.; Hedlund, E.; Fu, D.; Soldner, F.; Broccoli, V.; Constantine-Paton, M.; Isacson, O.; Jaenisch, R. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 5856–5861. [Google Scholar] [CrossRef]
- Ohta, S.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Kuwahara, R.; Ohyama, M.; Amagai, M.; Matsuzaki, Y.; Yamanaka, S.; Okano, H.; et al. Generation of human melanocytes from induced pluripotent stem cells. PLoS ONE 2011, 6, e16182. [Google Scholar] [CrossRef] [PubMed]
- Dell’Era, P.; Benzoni, P.; Crescini, E.; Valle, M.; Xia, E.; Consiglio, A.; Memo, M. Cardiac disease modeling using induced pluripotent stem cell-derived human cardiomyocytes. World J. Stem Cells 2015, 7, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Hirano, K.; Wu, S.M. iPS cell modeling of cardiometabolic diseases. J. Cardiovasc. Transl. Res. 2013, 6, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Tavian, D.; Missaglia, S.; Castagnetta, M.; Degiorgio, D.; Pennisi, E.M.; Coleman, R.A.; Dell’Era, P.; Mora, C.; Angelini, C.; Coviello, D.A. Generation of induced Pluripotent Stem Cells as disease modelling of NLSDM. Mol. Genet. Metab. 2017, 121, 28–34. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Missaglia, S.; Coleman, R.A.; Mordente, A.; Tavian, D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells 2019, 8, 187. https://doi.org/10.3390/cells8020187
Missaglia S, Coleman RA, Mordente A, Tavian D. Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells. 2019; 8(2):187. https://doi.org/10.3390/cells8020187
Chicago/Turabian StyleMissaglia, Sara, Rosalind A. Coleman, Alvaro Mordente, and Daniela Tavian. 2019. "Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function" Cells 8, no. 2: 187. https://doi.org/10.3390/cells8020187
APA StyleMissaglia, S., Coleman, R. A., Mordente, A., & Tavian, D. (2019). Neutral Lipid Storage Diseases as Cellular Model to Study Lipid Droplet Function. Cells, 8(2), 187. https://doi.org/10.3390/cells8020187