In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Cultures
2.3. Protein Content and Cell Size of HepaRG, PHH and HepG2
2.4. Karyotyping of HepaRG and HepG2 Cells
2.5. Immunocytochemistry Performed on HepaRG Cells
2.6. Mass Spectrometry Analyses Performed on the HepaRG Cell System
2.6.1. Protein Extraction
2.6.2. Sample Preparation for Intracellular Proteome Analysis
2.6.3. Sample Preparation for Secretome Analysis
2.6.4. Mass Spectrometry Analysis of the Intracellular Proteome
2.6.5. Mass Spectrometry Analysis of the Secretome
2.6.6. Mass Spectrometry Data Processing
2.6.7. Mass Spectrometry Data Analysis
2.6.8. Comparison of the Intracellular Proteome of the Three Cell Systems
2.6.9. Batch-to-Batch Stability of the Proteome of Differentiated HepaRG Cells (HPR116)
3. Results and Discussion
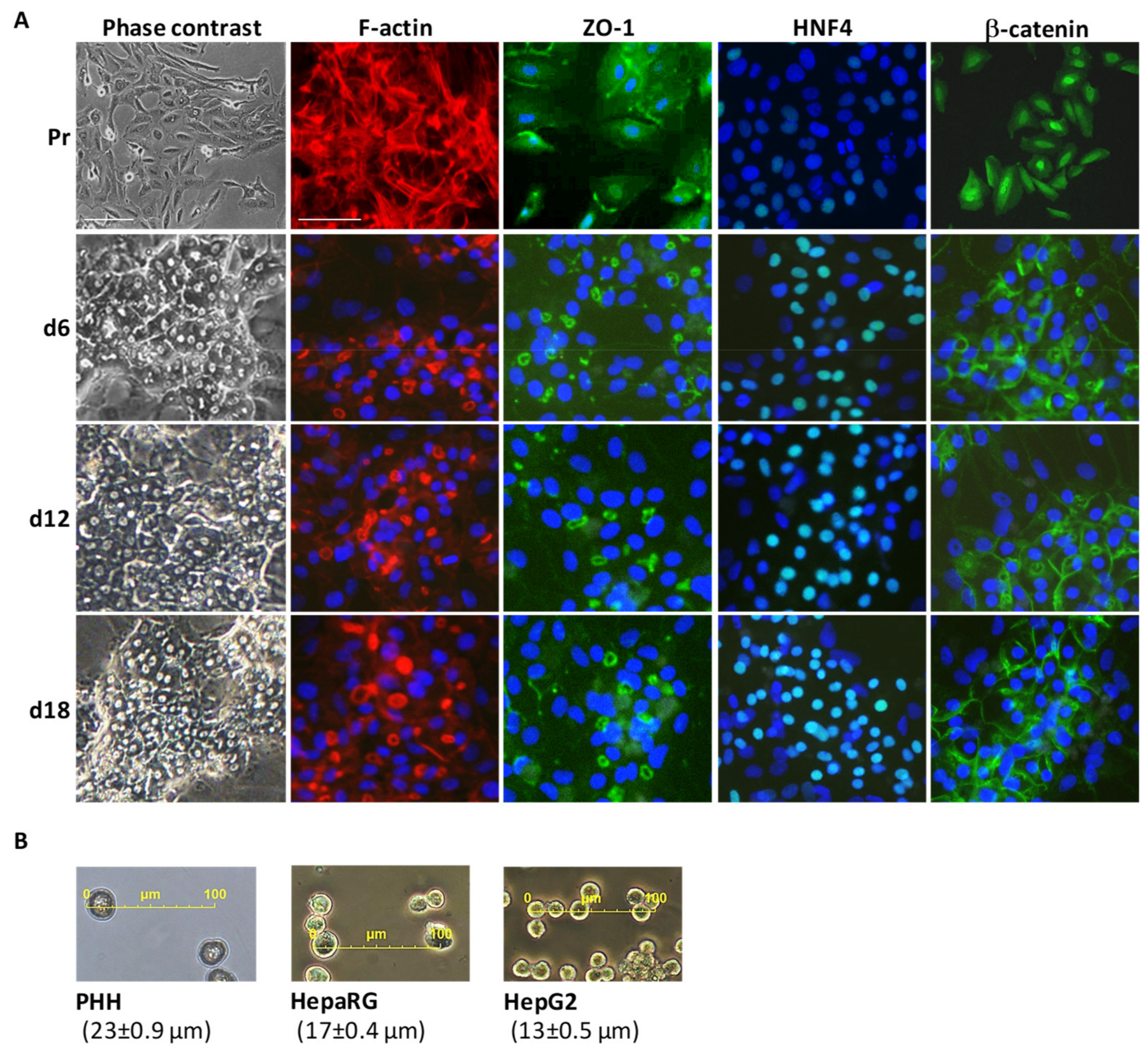
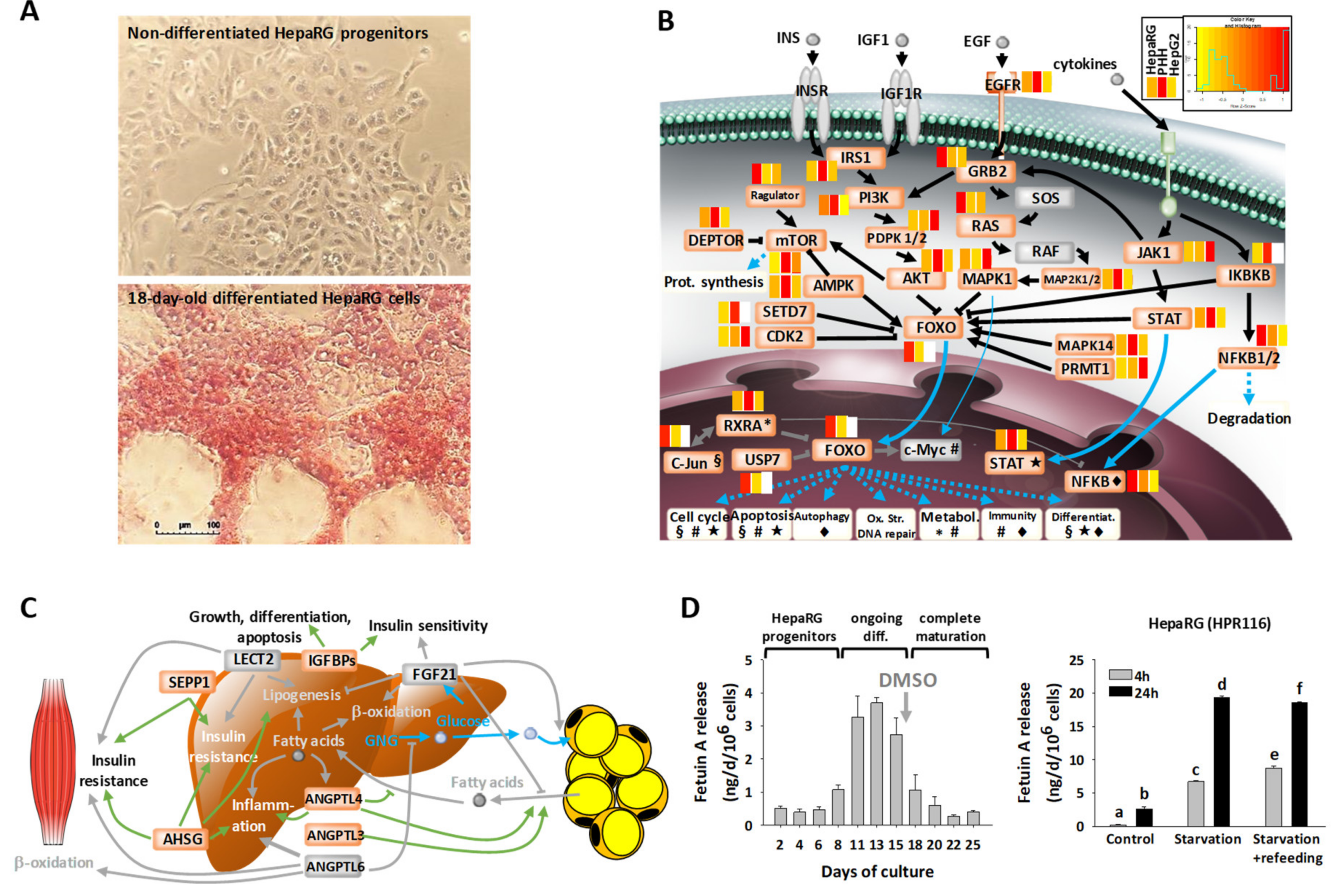
3.1. Main Characteristics of Highly Differentiated HepaRG Cells (HPR116)
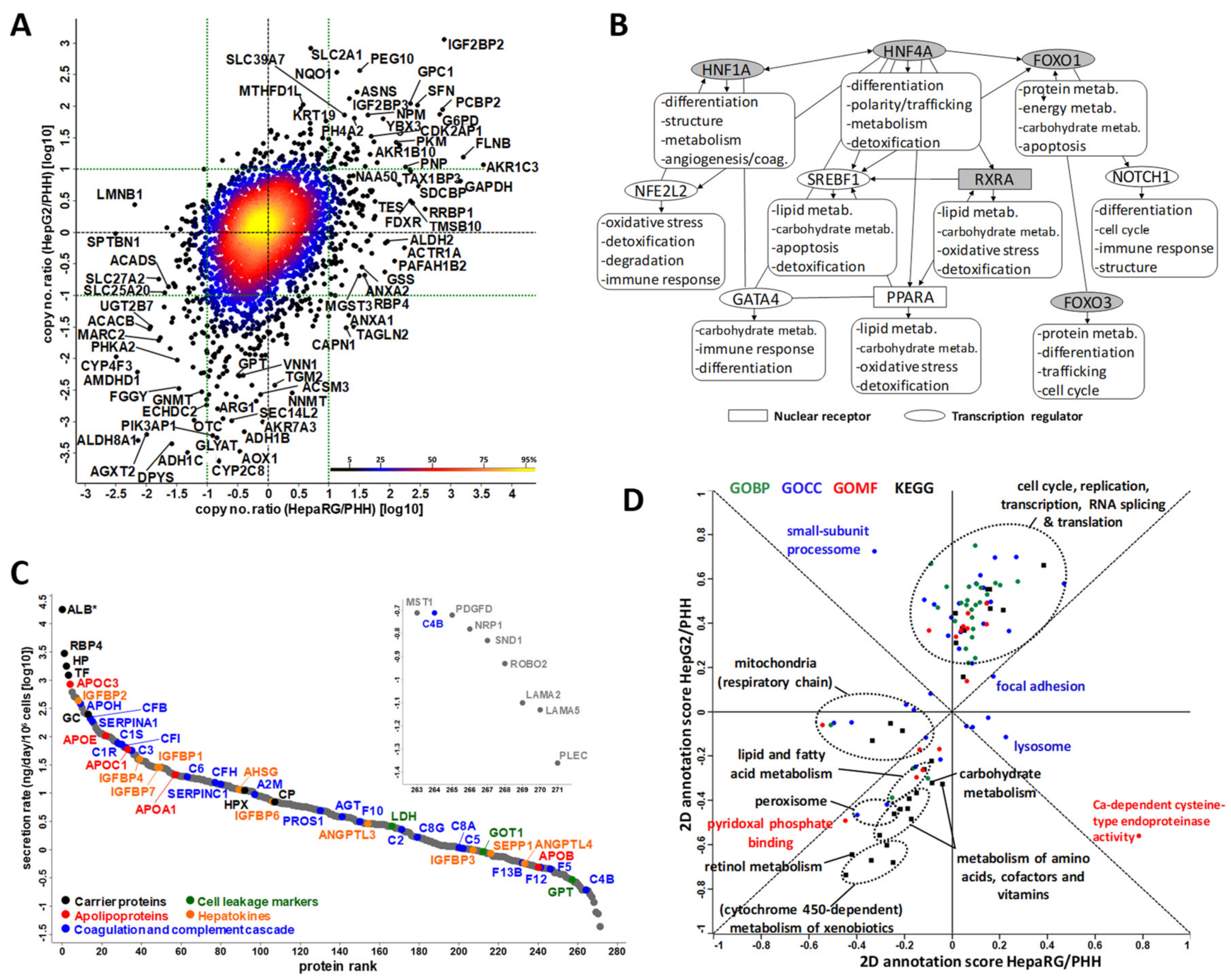
3.2. Large-Scale Proteomics of Highly Differentiated HepaRG
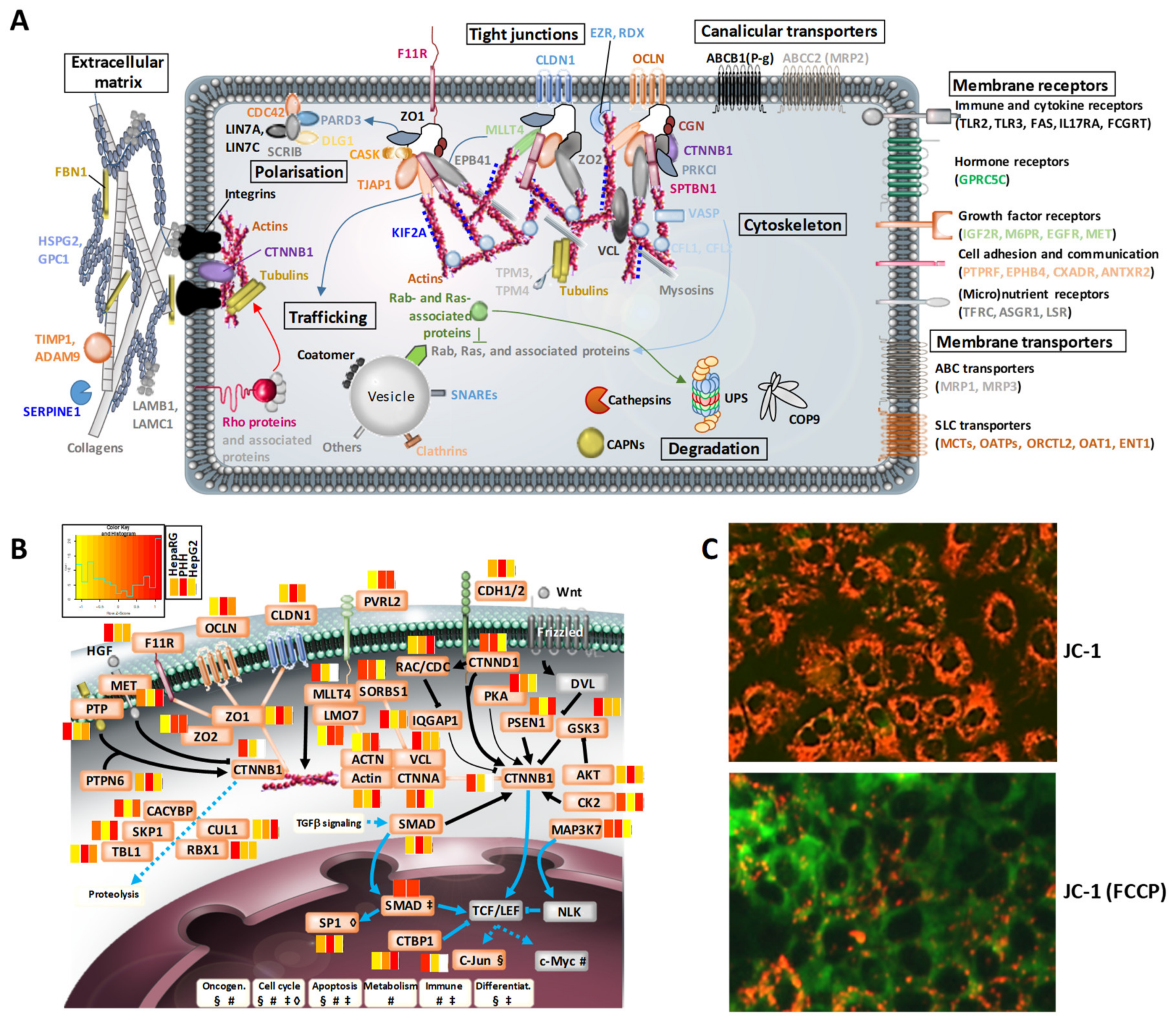
3.3. Structural Hotspots of Highly Differentiated HepaRG
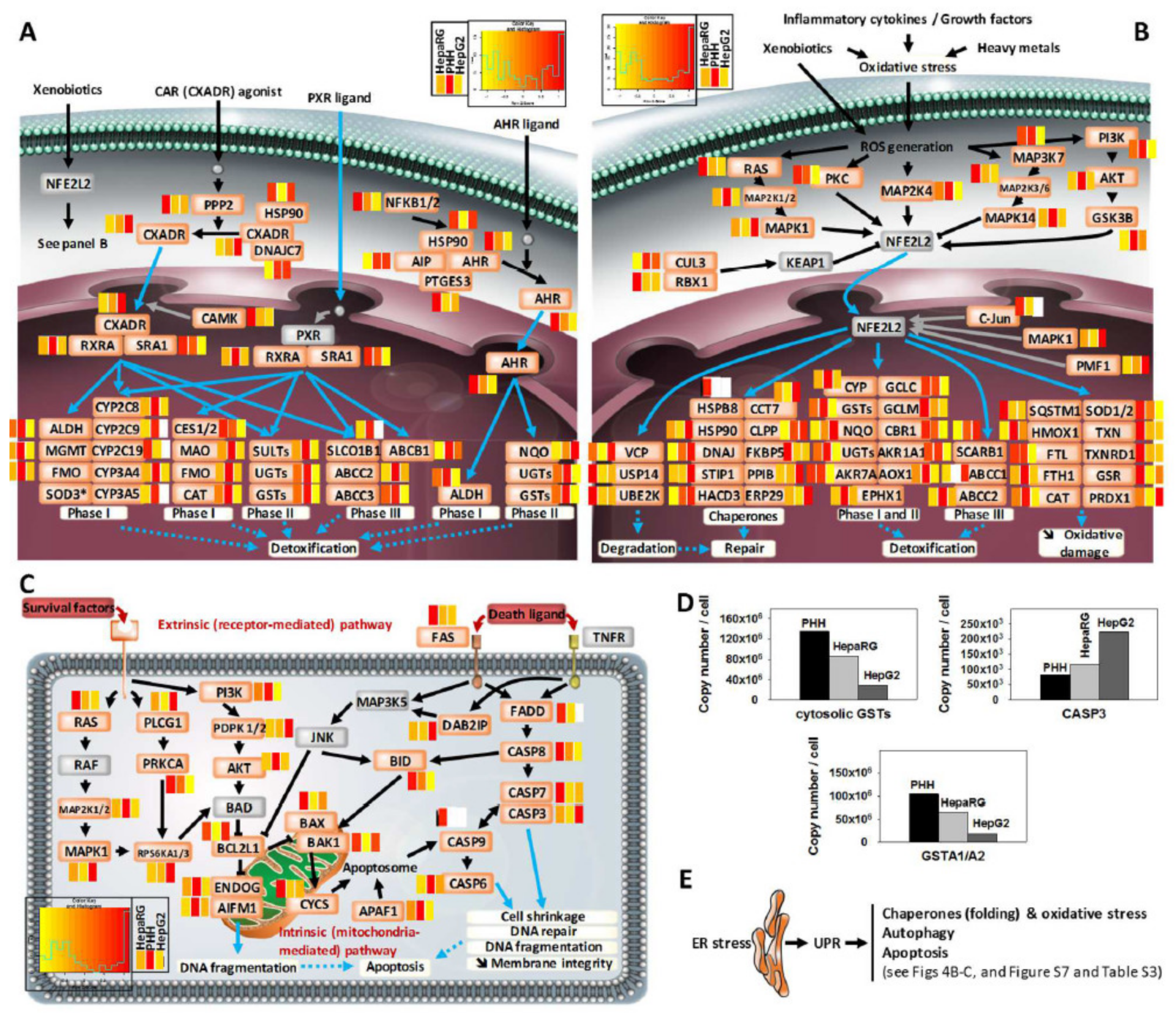
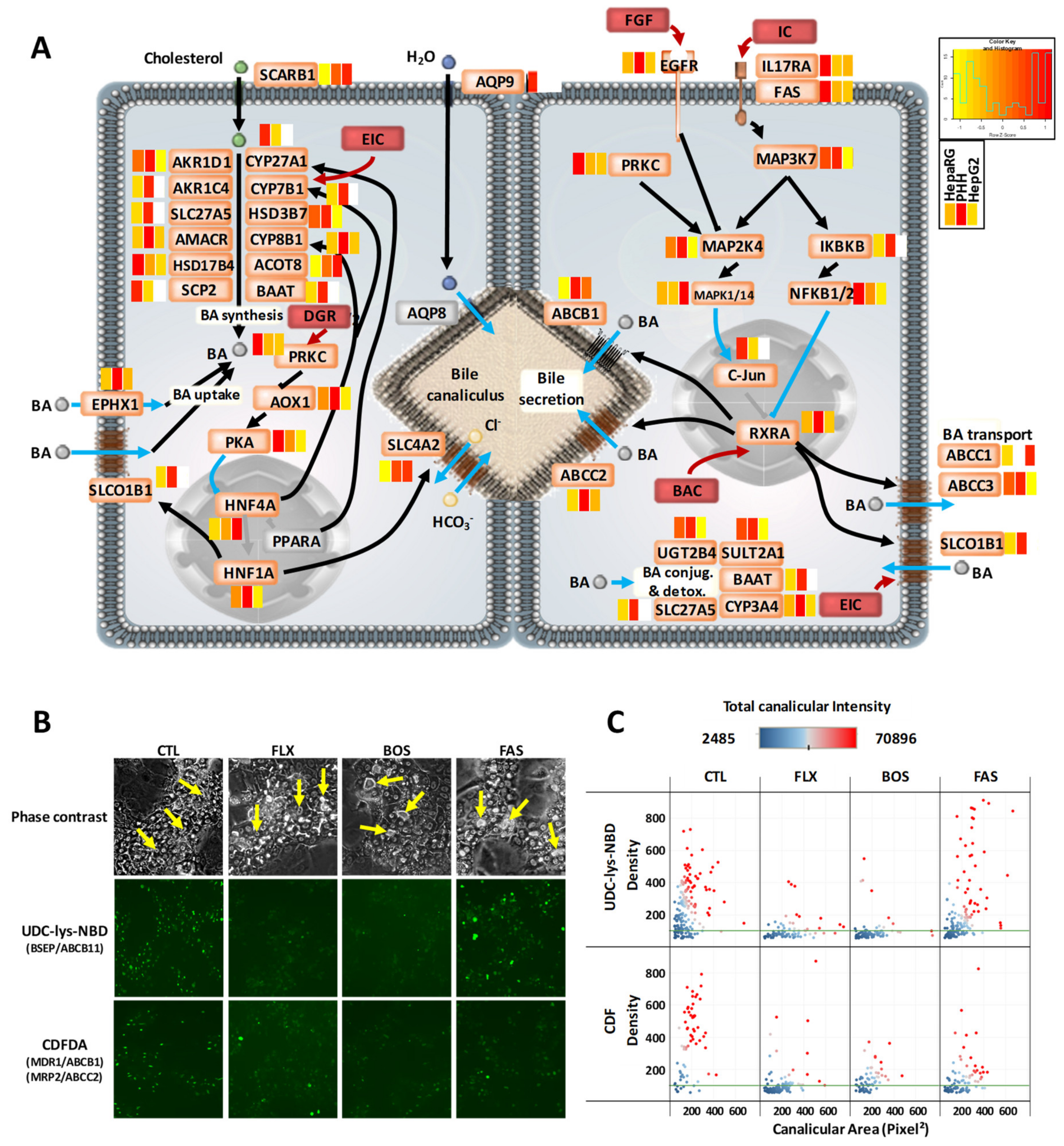
3.4. Comprehensive Detoxification Function of Highly Differentiated HepaRG Cells
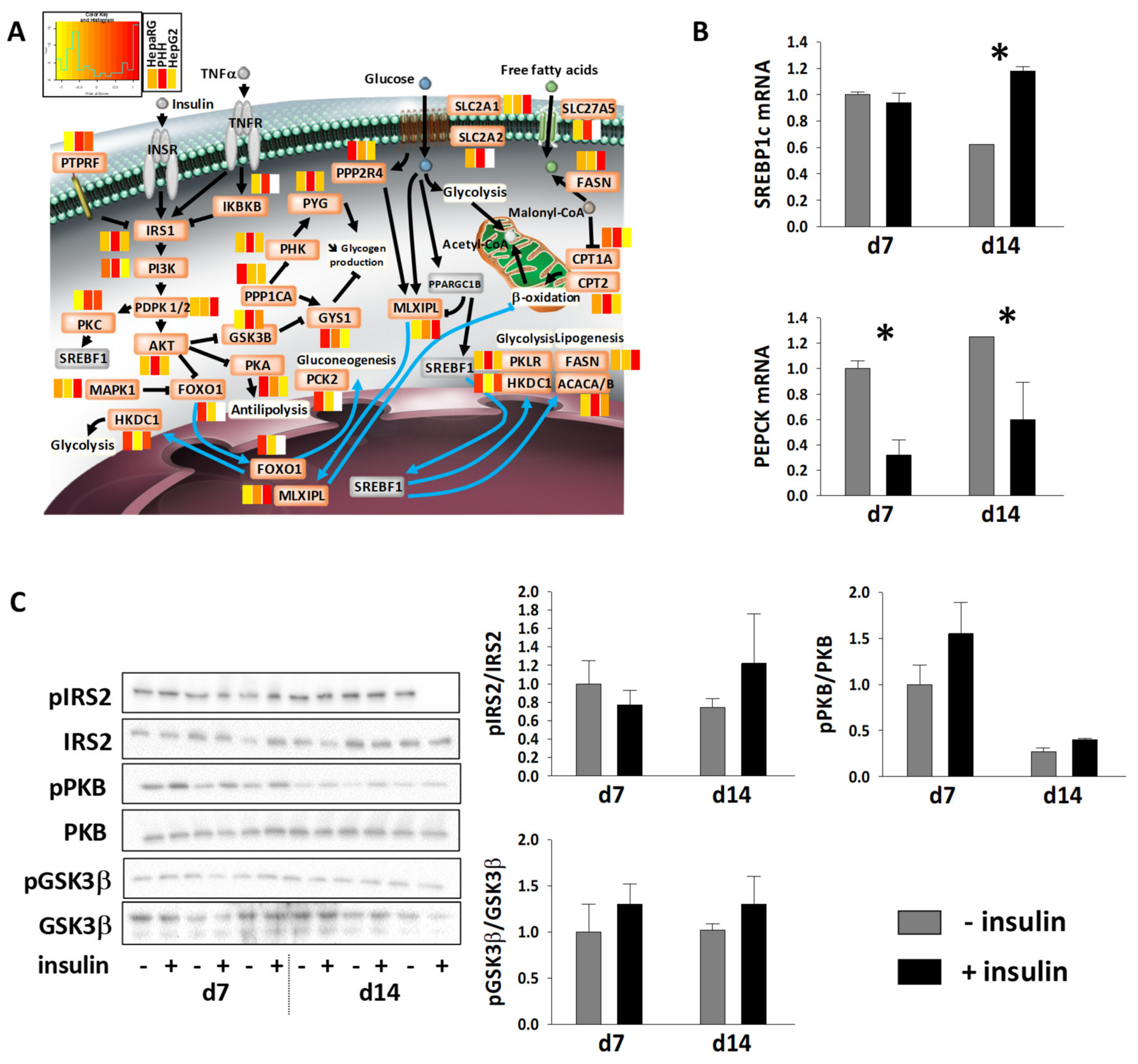
3.5. Energy Metabolic Functions of Highly Differentiated HepaRG
3.6. Disease-Related Pathways in Highly Differentiated HepaRG, a Promising Field of Application for the HepaRG Cell System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nagaki, M.; Moriwaki, H. Transcription factor HNF and hepatocyte differentiation. Hepatol Res. 2008, 38, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Guguen-Guillouzo, C.; Guillouzo, A. General review on in vitro hepatocyte models and their applications. Methods Mol. Biol. 2010, 640, 1–40. [Google Scholar] [PubMed]
- Godoy, P.; Widera, A.; Schmidt-Heck, W.; Campos, G.; Meyer, C.; Cadenas, C.; Reif, R.; Stober, R.; Hammad, S.; Putter, L.; et al. Gene network activity in cultivated primary hepatocytes is highly similar to diseased mammalian liver tissue. Arch. Toxicol. 2016, 90, 2513–2529. [Google Scholar] [CrossRef] [PubMed]
- Vildhede, A.; Wisniewski, J.R.; Noren, A.; Karlgren, M.; Artursson, P. Comparative Proteomic Analysis of Human Liver Tissue and Isolated Hepatocytes with a Focus on Proteins Determining Drug Exposure. J. Proteome Res. 2015, 14, 3305–3314. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Vildhede, A.; Noren, A.; Artursson, P. In-depth quantitative analysis and comparison of the human hepatocyte and hepatoma cell line HepG2 proteomes. J. Proteom. 2016, 136, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Matz-Soja, M. Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 2014, 20, 8491–8504. [Google Scholar] [CrossRef] [PubMed]
- Rogue, A.; Lambert, C.; Spire, C.; Claude, N.; Guillouzo, A. Interindividual variability in gene expression profiles in human hepatocytes and comparison with HepaRG cells. Drug Metab. Dispos. 2012, 40, 151–158. [Google Scholar] [CrossRef] [PubMed]
- LeCluyse, E.L. Human hepatocyte culture systems for the in vitro evaluation of cytochrome P450 expression and regulation. Eur. J. Pharm. Sci. 2001, 13, 343–368. [Google Scholar] [CrossRef]
- Andersson, T.B.; Kanebratt, K.P.; Kenna, J.G. The HepaRG cell line: A unique in vitro tool for understanding drug metabolism and toxicology in human. Expert Opin. Drug Metab. Toxicol. 2012, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Cerec, V.; Glaise, D.; Garnier, D.; Morosan, S.; Turlin, B.; Drenou, B.; Gripon, P.; Kremsdorf, D.; Guguen-Guillouzo, C.; Corlu, A. Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 2007, 45, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Lauschke, V.M.; Vorrink, S.U.; Palmgren, H.; Duffin, R.; Andersson, T.B.; Ingelman-Sundberg, M. Transcriptional, Functional, and Mechanistic Comparisons of Stem Cell-Derived Hepatocytes, HepaRG Cells, and Three-Dimensional Human Hepatocyte Spheroids as Predictive In Vitro Systems for Drug-Induced Liver Injury. Drug Metab. Dispos. 2017, 45, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Timmins-Schiffman, E.; Coton, M.; Coton, E.; Hymery, N.; Nunn, B.L.; Madec, S. Differential impacts of individual and combined exposures of deoxynivalenol and zearalenone on the HepaRG human hepatic cell proteome. J. Proteom. 2018, 173, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Rakus, D. Multi-enzyme digestion FASP and the ‘Total Protein Approach’-based absolute quantification of the Escherichia coli proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Mueller, D.; Schevchenko, V.; Noor, F. Long-term maintenance of HepaRG cells in serum-free conditions and application in a repeated dose study. J. Appl. Toxicol. 2014, 34, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Plumel, M.I.; Benhaim-Delarbre, M.; Rompais, M.; Thierse, D.; Sorci, G.; van Dorsselaer, A.; Criscuolo, F.; Bertile, F. Differential proteomics reveals age-dependent liver oxidative costs of innate immune activation in mice. J. Proteom. 2016, 135, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Uniprot. Available online: https://www.uniprot.org/ (accessed on 07 October 2016).
- Carapito, C.; Burel, A.; Guterl, P.; Walter, A.; Varrier, F.; Bertile, F.; Van Dorsselaer, A. MSDA, a proteomics software suite for in-depth Mass Spectrometry Data Analysis using grid computing. Proteomics 2014, 14, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: https://www.R-project.org/ (accessed on 15 September 2018).
- Chawade, A.; Alexandersson, E.; Levander, F. Normalyzer: A tool for rapid evaluation of normalization methods for omics data sets. J Proteome Res. 2014, 13, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W. Aneuploidy, polyploidy and ploidy reversal in the liver. Semin. Cell Dev. Biol. 2013, 24, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Ingenuity Pathway Analysis. Available online: https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ (accessed on 15 November 2016).
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: https://www.genome.jp/kegg/ (accessed on 30 April 2018).
- SignalP-4.1 Server. Available online: http://www.cbs.dtu.dk/services/SignalP/ (accessed on 15 November 2016).
- SecretomeP 2.0 Server. Available online: http://www.cbs.dtu.dk/services/SecretomeP/ (accessed on 15 November 2016).
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. 1D and 2D annotation enrichment: A statistical method integrating quantitative proteomics with complementary high-throughput data. BMC Bioinform. 2012, 13 (Suppl. 16), S12. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.V.; Piersma, S.R.; Warmoes, M.; Jimenez, C.R. On the beta-binomial model for analysis of spectral count data in label-free tandem mass spectrometry-based proteomics. Bioinformatics 2010, 26, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.P.; Li, L.; Chamberlain, E.D.; Wang, H.; Ferguson, S.S. Contextualizing Hepatocyte Functionality of Cryopreserved HepaRG Cell Cultures. Drug Metab. Dispos. 2016, 44, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Knowles, B.B.; Howe, C.C.; Aden, D.P. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980, 209, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Terrada, D.; Cheung, S.W.; Finegold, M.J.; Knowles, B.B. Hep G2 is a hepatoblastoma-derived cell line. Hum. Pathol. 2009, 40, 1512–1515. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. What is the total number of protein molecules per cell volume? A call to rethink some published values. Bioessays 2013, 35, 1050–1055. [Google Scholar] [CrossRef] [PubMed]
- Gissen, P.; Arias, I.M. Structural and functional hepatocyte polarity and liver disease. J. Hepatol. 2015, 63, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Sharanek, A.; Burban, A.; Burbank, M.; Le Guevel, R.; Li, R.; Guillouzo, A.; Guguen-Guillouzo, C. Rho-kinase/myosin light chain kinase pathway plays a key role in the impairment of bile canaliculi dynamics induced by cholestatic drugs. Sci. Rep. 2016, 6, 24709. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Thenappan, A.; Li, Y.; Kitisin, K.; Rashid, A.; Shetty, K.; Johnson, L.; Mishra, L. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology 2010, 51, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Guillemot, L.; Schneider, Y.; Brun, P.; Castagliuolo, I.; Pizzuti, D.; Martines, D.; Jond, L.; Bongiovanni, M.; Citi, S. Cingulin is dispensable for epithelial barrier function and tight junction structure, and plays a role in the control of claudin-2 expression and response to duodenal mucosa injury. J. Cell Sci. 2012, 125, 5005–5014. [Google Scholar] [CrossRef] [PubMed]
- Russel, J.O.; Monga, S.P. Wnt/β-catenin signaling in liver development, homeostasis, and pathobiology. Annu. Rev. Pathol. Mech. Dis. 2018, 13, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Terrada, D.; Gunaratne, P.H.; Adesina, A.M.; Pulliam, J.; Hoang, D.M.; Nguyen, Y.; Mistretta, T.A.; Margolin, J.; Finegold, M.J. Histologic subtypes of hepatoblastoma are characterized by differential canonical Wnt and Notch pathway activation in DLK+ precursors. Hum. Pathol. 2009, 40, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Torre, C.; Perret, C.; Colnot, S. Transcription dynamics in a physiological process: Beta-catenin signaling directs liver metabolic zonation. Int. J. Biochem. Cell Biol. 2011, 43, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, R.; Nibourg, G.A.; van der Hoeven, T.V.; Ackermans, M.T.; Hakvoort, T.B.; van Gulik, T.M.; Lamers, W.H.; Elferink, R.P.; Chamuleau, R.A. The HepaRG cell line is suitable for bioartificial liver application. Int. J. Biochem. Cell Biol. 2011, 43, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Moedas, M.F.; Adam, A.A.A.; Farelo, M.A.; IJlst, L.; Chalumeau, R.A.F.M.; Hoekstra, R.; Wanders, R.J.A.; Silva, M.F.B. Advances in methods for characterization of hepatic urea cycle enzymatic activity in HepaRG cells using UPLC-MS/MS. Anal. Biochem. 2017, 535, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Pernelle, K.; Le Guevel, R.; Glaise, D.; Stasio, C.G.; Le Charpentier, T.; Bouaita, B.; Corlu, A.; Guguen-Guillouzo, C. Automated detection of hepatotoxic compounds in human hepatocytes using HepaRG cells and image-based analysis of mitochondrial dysfunction with JC-1 dye. Toxicol. Appl. Pharm. 2011, 254, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Li, C.Y.; Kong, A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch. Pharm. Res. 2005, 28, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Aninat, C.; Piton, A.; Glaise, D.; Le Charpentier, T.; Langouet, S.; Morel, F.; Guguen-Guillouzo, C.; Guillouzo, A. Expression of cytochromes P450, conjugating enzymes and nuclear receptors in human hepatoma HepaRG cells. Drug Metab. Dispos. 2006, 34, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Wigger, L.; Casals-Casas, C.; Baruchet, M.; Trang, K.; Pradervand, S.; Naldi, A.; Desvergne, B. System analysis of the functional cross-talk between PPARalpha, LXR and FXR in the human HepaRG liver cells. bioRxiv 2019, 514976. [Google Scholar] [CrossRef]
- Kanebratt, K.P.; Andersson, T.B. Evaluation of HepaRG cells as an in vitro model for human drug metabolism studies. Drug Metab. Dispos. 2008, 36, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Attignon, E.A.; Leblanc, A.F.; Le-Grand, B.; Duval, C.; Aggerbeck, M.; Rouach, H.; Blanc, E.B. Novel roles for AhR and ARNT in the regulation of alcohol dehydrogenases in human hepatic cells. Arch. Toxicol. 2017, 91, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Kalen, A.L.; Goswami, P.C. Manganese superoxide dismutase regulates a redox cycle within the cell cycle. Antioxid. Redox Signal. 2014, 20, 1618–1627. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zou, Y.; Nambiar, S.M.; Lee, J.; Yang, Y.; Dai, G. Keap1 modulates the redox cycle and hepatocyte cell cycle in regenerating liver. Cell Cycle 2014, 13, 2349–2358. [Google Scholar] [CrossRef] [PubMed]
- Gilot, D.; Loyer, P.; Corlu, A.; Glaise, D.; Lagadic-Gossmann, D.; Atfi, A.; Morel, F.; Ichijo, H.; Guguen-Guillouzo, C. Liver protection from apoptosis requires both blockage of initiator caspase activities and inhibition of ASK1/JNK pathway via glutathione S-transferase regulation. J. Biol. Chem. 2002, 277, 49220–49229. [Google Scholar] [CrossRef] [PubMed]
- Burban, A.; Sharanek, A.; Hue, R.; Gay, M.; Routier, S.; Guillouzo, A.; Guguen-Guillouzo, C. Penicillinase-resistant antibiotics induce non-immune-mediated cholestasis through HSP27 activation associated with PKC/P38 and PI3K/AKT signaling pathways. Sci. Rep. 2017, 7, 1815. [Google Scholar] [CrossRef] [PubMed]
- Madec, S.; Cerec, V.; Plee-Gautier, E.; Antoun, J.; Glaise, D.; Salaun, J.P.; Guguen-Guillouzo, C.; Corlu, A. CYP4F3B expression is associated with differentiation of HepaRG human hepatocytes and unaffected by fatty acid overload. Drug Metab. Dispos. 2011, 39, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Samanez, C.H.; Caron, S.; Briand, O.; Dehondt, H.; Duplan, I.; Kuipers, F.; Hennuyer, H.; Clavey, V.; Staels, B. The human hepatocyte cell lines IHH and HepaRG: Models to study glucose, lipid and lipoprotein metabolism. Arch. Physiol. Biochem. 2012, 118, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.A.; Tabata, M.; Rodgers, J.T.; Puigserver, P. USP7 attenuates hepatic gluconeogenesis through modulation of FoxO1 gene promoter occupancy. Mol. Endocrinol. 2014, 28, 912–924. [Google Scholar] [CrossRef] [PubMed]
- Adachi, M.; Osawa, Y.; Uchinami, H.; Kitamura, T.; Accili, D.; Brenner, D.A. The forkhead transcription factor FoxO1 regulates proliferation and transdifferentiation of hepatic stellate cells. Gastroenterology 2007, 132, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Parent, R.; Kolippakkam, D.; Booth, G.; Beretta, L. Mammalian target of rapamycin activation impairs hepatocytic differentiation and targets genes moderating lipid homeostasis and hepatocellular growth. Cancer Res. 2007, 67, 4337–4345. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Reynolds, J.; Chew, S.; Lamba, D.A.; Hughes, R.E. DEPTOR is a stemness factor that regulates pluripotency of embryonic stem cells. J. Biol. Chem. 2014, 289, 31818–31826. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Haring, H.U. The role of hepatokines in metabolism. Nat. Rev. Endocrinol. 2013, 9, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.Y.; Wu, H.T.; Hung, H.C.; Yang, Y.C.; Wu, J.S.; Chang, C.J. Endoplasmic reticulum stress induces the expression of fetuin-A to develop insulin resistance. Endocrinology 2012, 153, 2974–2984. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.; Trauner, M. Recent advances in understanding and managing cholestasis. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ko, K.; Xia, M.; Li, T.W.; Oh, P.; Li, J.; Lu, S.C. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology 2010, 51, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Sharanek, A.; Azzi, P.B.; Al-Attrache, H.; Savary, C.C.; Humbert, L.; Rainteau, D.; Guguen-Guillouzo, C.; Guillouzo, A. Different dose-dependent mechanisms are involved in early cyclosporine a-induced cholestatic effects in hepaRG cells. Toxicol. Sci. 2014, 141, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Dessalle, K.; Euthine, V.; Chanon, S.; Delarichaudy, J.; Fujii, I.; Rome, S.; Vidal, H.; Nemoz, G.; Simon, C.; Lefai, E. SREBP-1 transcription factors regulate skeletal muscle cell size by controlling protein synthesis through myogenic regulatory factors. PLoS One 2012, 7, e50878. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tascher, G.; Burban, A.; Camus, S.; Plumel, M.; Chanon, S.; Le Guevel, R.; Shevchenko, V.; Van Dorsselaer, A.; Lefai, E.; Guguen-Guillouzo, C.; et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells 2019, 8, 192. https://doi.org/10.3390/cells8020192
Tascher G, Burban A, Camus S, Plumel M, Chanon S, Le Guevel R, Shevchenko V, Van Dorsselaer A, Lefai E, Guguen-Guillouzo C, et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells. 2019; 8(2):192. https://doi.org/10.3390/cells8020192
Chicago/Turabian StyleTascher, Georg, Audrey Burban, Sandrine Camus, Marine Plumel, Stéphanie Chanon, Remy Le Guevel, Valery Shevchenko, Alain Van Dorsselaer, Etienne Lefai, Christiane Guguen-Guillouzo, and et al. 2019. "In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes" Cells 8, no. 2: 192. https://doi.org/10.3390/cells8020192
APA StyleTascher, G., Burban, A., Camus, S., Plumel, M., Chanon, S., Le Guevel, R., Shevchenko, V., Van Dorsselaer, A., Lefai, E., Guguen-Guillouzo, C., & Bertile, F. (2019). In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells, 8(2), 192. https://doi.org/10.3390/cells8020192