Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease


Abstract
:1. Introduction
2. Diet as A Risk Factor for IBD: Epidemiological Studies
3. Diet as A Modulator of Gut Microbiota and Their Metabolites
4. GPCRs Sense Microbial-Derived Metabolites
5. Metabolites and Mucosal Inflammation
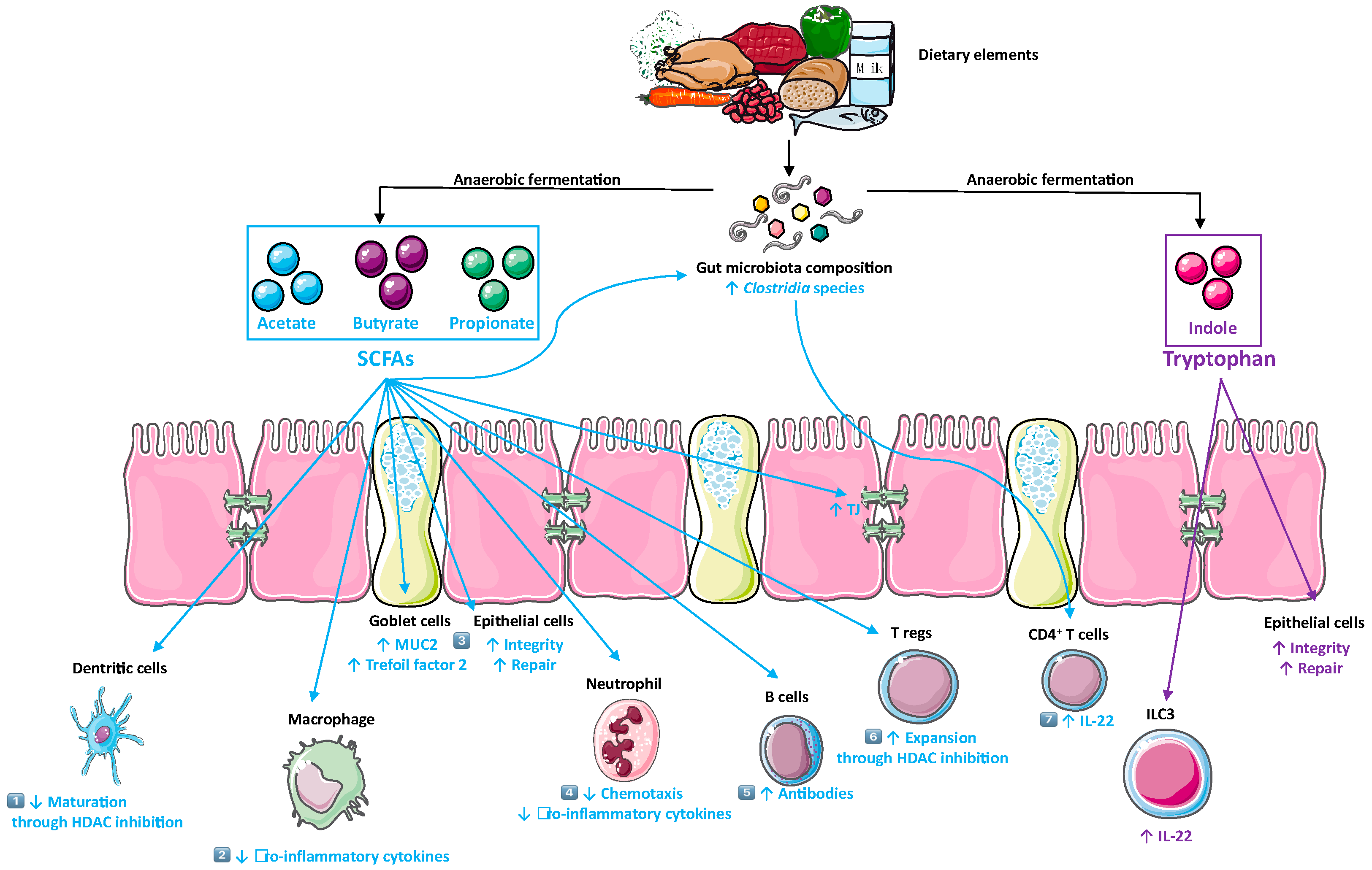
5.1. SCFAs
5.2. Tryptophan Metabolites
6. Metabolite-Sensing G Protein-Coupled Receptors
6.1. GPCRs for Short Chain Fatty Acids (SCFAs)
6.2. GPCRs for Medium and Long Chain Fatty Acid (MCFA, LCFA)
6.3. GPCRs for Amino Acids and Related Metabolites
6.4. Bile Acid-Sensing GPCRs
6.5. pH-Sensitive Receptor
6.6. GPCRs for Citric Acid Cycle Intermediates
7. Metabolite-Sensing GPCRs and Inflammatory Bowel Disease
8. Possible Mechanisms whereby Metabolite-Sensing GPCRs Exerts an Anti-Inflammatory Effect in the Gut
8.1. Metabolite-Sensing GPCRs Enhance Epithelial Integrity
8.2. Metabolite-Sensing GPCRs Promote the Intestinal Immune System
9. Potential Tools for Studying Diet and Bacterial-Derived Metabolites in the Context of IBD
10. Metabolite-Sensing GPCRs for the Treatment of IBD: What Are the Challenges?
11. Conclusions
Funding
Conflicts of Interest
Abbreviations
References
- Strober, W.; Fuss, I.; Mannon, P. The fundamental basis of inflammatory bowel disease. J. Clin. Investig. 2007, 117, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, D.C.; Sandborn, W.J. Inflammatory bowel disease: Clinical aspects and established and evolving therapies. Lancet 2007, 369, 1641–1657. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Soon, I.S.; Rabi, D.M.; Ghali, W.A.; Ferris, M.; Chernoff, G.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Barkema, H.W.; et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 2012, 142, 46–54. [Google Scholar] [CrossRef]
- Ng, S.C.; Bernstein, C.N.; Vatn, M.H.; Lakatos, P.L.; Loftus, E.V., Jr.; Tysk, C.; O’Morain, C.; Moum, B.; Colombel, J.F.; Epidemiology; et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 2013, 62, 630–649. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Korzenik, J.R.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology 2013, 145, 970–977. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; Siuzdak, G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [Green Version]
- Rasoamanana, R.; Darcel, N.; Fromentin, G.; Tome, D. Nutrient sensing and signalling by the gut. Proc. Nutr. Soc. 2012, 71, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Curto, E.; Milligan, G. Metabolism meets immunity: The role of free fatty acid receptors in the immune system. Biochem. Pharm. 2016, 114, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef]
- Uchimura, Y.; Fuhrer, T.; Li, H.; Lawson, M.A.; Zimmermann, M.; Yilmaz, B.; Zindel, J.; Ronchi, F.; Sorribas, M.; Hapfelmeier, S.; et al. Antibodies Set Boundaries Limiting Microbial Metabolite Penetration and the Resultant Mammalian Host Response. Immunity 2018, 49, 545–559.e5. [Google Scholar] [CrossRef]
- Tan, J.K.; McKenzie, C.; Marino, E.; Macia, L.; Mackay, C.R. Metabolite-Sensing G Protein-Coupled Receptors-Facilitators of Diet-Related Immune Regulation. Annu. Rev. Immunol. 2017, 35, 371–402. [Google Scholar] [CrossRef]
- Ng, S.C.; Kaplan, G.G.; Tang, W.; Banerjee, R.; Adigopula, B.; Underwood, F.E.; Tanyingoh, D.; Wei, S.C.; Lin, W.C.; Lin, H.H.; et al. Population Density and Risk of Inflammatory Bowel Disease: A Prospective Population-Based Study in 13 Countries or Regions in Asia-Pacific. Am. J. Gastroenterol. 2019, 114, 107–115. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Bernstein, C.N.; Iliopoulos, D.; Macpherson, A.; Neurath, M.F.; Ali, R.A.R.; Vavricka, S.R.; Fiocchi, C. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 39–49. [Google Scholar] [CrossRef]
- Lee, D.; Albenberg, L.; Compher, C.; Baldassano, R.; Piccoli, D.; Lewis, J.D.; Wu, G.D. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology 2015, 148, 1087–1106. [Google Scholar] [CrossRef]
- Khalili, H.; Malik, S.; Ananthakrishnan, A.N.; Garber, J.J.; Higuchi, L.M.; Joshi, A.; Peloquin, J.; Richter, J.M.; Stewart, K.O.; Curhan, G.C.; et al. Identification and Characterization of a Novel Association between Dietary Potassium and Risk of Crohn’s Disease and Ulcerative Colitis. Front. Immunol. 2016, 7, 554. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Song, M.; Higuchi, L.M.; Richter, J.M.; Chan, A.T. Zinc intake and risk of Crohn’s disease and ulcerative colitis: A prospective cohort study. Int. J. Epidemiol. 2015, 44, 1995–2005. [Google Scholar] [CrossRef]
- Investigators, I.B.D.i.E.S.; Tjonneland, A.; Overvad, K.; Bergmann, M.M.; Nagel, G.; Linseisen, J.; Hallmans, G.; Palmqvist, R.; Sjodin, H.; Hagglund, G.; et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: A nested case-control study within a European prospective cohort study. Gut 2009, 58, 1606–1611. [Google Scholar] [CrossRef]
- Fallani, M.; Young, D.; Scott, J.; Norin, E.; Amarri, S.; Adam, R.; Aguilera, M.; Khanna, S.; Gil, A.; Edwards, C.A.; et al. Intestinal microbiota of 6-week-old infants across Europe: Geographic influence beyond delivery mode, breast-feeding, and antibiotics. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 77–84. [Google Scholar] [CrossRef]
- Nabhani, Z. A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity 2019. [Google Scholar] [CrossRef]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Klingbeil, E.; de La Serre, C.B. Microbiota modulation by eating patterns and diet composition: Impact on food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R1254–R1260. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, J.; Lange, B.; Frick, J.S.; Sauer, H.; Zimmermann, K.; Schwiertz, A.; Rusch, K.; Klosterhalfen, S.; Enck, P. A vegan or vegetarian diet substantially alters the human colonic faecal microbiota. Eur. J. Clin. Nutr. 2012, 66, 53–60. [Google Scholar] [CrossRef] [PubMed]
- N, I.J.; Derrien, M.; van Doorn, G.M.; Rijnierse, A.; van den Bogert, B.; Muller, M.; Dekker, J.; Kleerebezem, M.; van der Meer, R. Dietary heme alters microbiota and mucosa of mouse colon without functional changes in host-microbe cross-talk. PLoS ONE 2012, 7, e49868. [Google Scholar] [CrossRef]
- Ijssennagger, N.; Belzer, C.; Hooiveld, G.J.; Dekker, J.; van Mil, S.W.; Muller, M.; Kleerebezem, M.; van der Meer, R. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc. Natl. Acad. Sci. USA 2015, 112, 10038–10043. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Marcobal, A.; Kashyap, P.C.; Nelson, T.A.; Aronov, P.A.; Donia, M.S.; Spormann, A.; Fischbach, M.A.; Sonnenburg, J.L. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. ISME J. 2013, 7, 1933–1943. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.H.; Macfarlane, G.T. Role of intestinal bacteria in nutrient metabolism. JPEN J. Parenter Enteral. Nutr. 1997, 21, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Topping, D.L.; Clifton, P.M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, S.; Cochrane, S. Alteration of sulfate and hydrogen metabolism in the human colon by changing intestinal transit rate. Am. J. Gastroenterol. 2007, 102, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.; Duncan, S.H.; Holtrop, G.; Anderson, S.E.; Lobley, G.E.; Flint, H.J. Impact of pH on lactate formation and utilization by human fecal microbial communities. Appl. Environ. Microbiol. 2007, 73, 6526–6533. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Eyster, K.M. The membrane and lipids as integral participants in signal transduction: Lipid signal transduction for the non-lipid biochemist. Adv. Physiol. Educ. 2007, 31, 5–16. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef]
- Huda-Faujan, N.; Abdulamir, A.S.; Fatimah, A.B.; Anas, O.M.; Shuhaimi, M.; Yazid, A.M.; Loong, Y.Y. The impact of the level of the intestinal short chain Fatty acids in inflammatory bowel disease patients versus healthy subjects. Open Biochem. J. 2010, 4, 53–58. [Google Scholar] [CrossRef]
- Treem, W.R.; Ahsan, N.; Shoup, M.; Hyams, J.S. Fecal short-chain fatty acids in children with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 1994, 18, 159–164. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Thibault, R.; De Coppet, P.; Daly, K.; Bourreille, A.; Cuff, M.; Bonnet, C.; Mosnier, J.F.; Galmiche, J.P.; Shirazi-Beechey, S.; Segain, J.P. Down-regulation of the monocarboxylate transporter 1 is involved in butyrate deficiency during intestinal inflammation. Gastroenterology 2007, 133, 1916–1927. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Jenq, R.; Mathew, A.V.; Koenigsknecht, M.; Hanash, A.; Toubai, T.; Oravecz-Wilson, K.; Wu, S.R.; Sun, Y.; Rossi, C.; et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat. Immunol. 2016, 17, 505–513. [Google Scholar] [CrossRef]
- Harig, J.M.; Soergel, K.H.; Komorowski, R.A.; Wood, C.M. Treatment of diversion colitis with short-chain-fatty acid irrigation. N. Engl. J. Med. 1989, 320, 23–28. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 1992, 103, 51–56. [Google Scholar] [CrossRef]
- Breuer, R.I.; Buto, S.K.; Christ, M.L.; Bean, J.; Vernia, P.; Paoluzi, P.; Di Paolo, M.C.; Caprilli, R. Rectal irrigation with short-chain fatty acids for distal ulcerative colitis. Preliminary report. Dig. Dis. Sci. 1991, 36, 185–187. [Google Scholar] [CrossRef]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFβ1 signalling in inflammation. Mucosal. Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef]
- Luhrs, H.; Gerke, T.; Muller, J.G.; Melcher, R.; Schauber, J.; Boxberge, F.; Scheppach, W.; Menzel, T. Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [Green Version]
- Gaudier, E.; Jarry, A.; Blottiere, H.M.; de Coppet, P.; Buisine, M.P.; Aubert, J.P.; Laboisse, C.; Cherbut, C.; Hoebler, C. Butyrate specifically modulates MUC gene expression in intestinal epithelial goblet cells deprived of glucose. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1168–G1174. [Google Scholar] [CrossRef]
- Hatayama, H.; Iwashita, J.; Kuwajima, A.; Abe, T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem. Biophys. Res. Commun. 2007, 356, 599–603. [Google Scholar] [CrossRef]
- Correa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A. Regulation of immune cell function by short-chain fatty acids. Clin. Transl. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [Green Version]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.E.; Inglis, G.D. Butyrate Supplementation at High Concentrations Alters Enteric Bacterial Communities and Reduces Intestinal Inflammation in Mice Infected with Citrobacter rodentium. mSphere 2017, 2. [Google Scholar] [CrossRef]
- Gaudier, E.; Rival, M.; Buisine, M.P.; Robineau, I.; Hoebler, C. Butyrate enemas upregulate Muc genes expression but decrease adherent mucus thickness in mice colon. Physiol. Res. 2009, 58, 111–119. [Google Scholar]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef]
- Kaisar, M.M.M.; Pelgrom, L.R.; van der Ham, A.J.; Yazdanbakhsh, M.; Everts, B. Butyrate Conditions Human Dendritic Cells to Prime Type 1 Regulatory T Cells via both Histone Deacetylase Inhibition and G Protein-Coupled Receptor 109A Signaling. Front. Immunol. 2017, 8, 1429. [Google Scholar] [CrossRef]
- Nastasi, C.; Fredholm, S.; Willerslev-Olsen, A.; Hansen, M.; Bonefeld, C.M.; Geisler, C.; Andersen, M.H.; Odum, N.; Woetmann, A. Butyrate and propionate inhibit antigen-specific CD8(+) T cell activation by suppressing IL-12 production by antigen-presenting cells. Sci. Rep. 2017, 7, 14516. [Google Scholar] [CrossRef]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Li, L.; Min, J.; Wang, J.; Wu, H.; Zeng, Y.; Chen, S.; Chu, Z. Butyrate interferes with the differentiation and function of human monocyte-derived dendritic cells. Cell Immunol. 2012, 277, 66–73. [Google Scholar] [CrossRef]
- Millard, A.L.; Mertes, P.M.; Ittelet, D.; Villard, F.; Jeannesson, P.; Bernard, J. Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin. Exp. Immunol. 2002, 130, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Qie, Y.; Park, J.; Kim, C.H. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe 2016, 20, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Geuking, M.B.; Cahenzli, J.; Lawson, M.A.; Ng, D.C.; Slack, E.; Hapfelmeier, S.; McCoy, K.D.; Macpherson, A.J. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011, 34, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, C.; Scully, P.; O’Mahony, D.; Murphy, S.; O’Brien, F.; Lyons, A.; Sherlock, G.; MacSharry, J.; Kiely, B.; Shanahan, F.; et al. Commensal-induced regulatory T cells mediate protection against pathogen-stimulated NF-kappaB activation. PLoS Pathog. 2008, 4, e1000112. [Google Scholar] [CrossRef]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaus, S.; Schulte, B.; Al-Massad, N.; Thieme, F.; Schulte, D.M.; Bethge, J.; Rehman, A.; Tran, F.; Aden, K.; Hasler, R.; et al. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology 2017, 153, 1504–1516.e2. [Google Scholar] [CrossRef] [PubMed]
- Gurtner, G.J.; Newberry, R.D.; Schloemann, S.R.; McDonald, K.G.; Stenson, W.F. Inhibition of indoleamine 2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in mice. Gastroenterology 2003, 125, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef]
- Lee, J.S.; Cella, M.; McDonald, K.G.; Garlanda, C.; Kennedy, G.D.; Nukaya, M.; Mantovani, A.; Kopan, R.; Bradfield, C.A.; Newberry, R.D.; et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat. Immunol. 2011, 13, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Faith, N.G.; Nakayama, Y.; Suresh, M.; Steinberg, H.; Czuprynski, C.J. The aryl hydrocarbon receptor is required for optimal resistance to Listeria monocytogenes infection in mice. J. Immunol. 2007, 179, 6952–6962. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Kiss, E.A.; Vonarbourg, C.; Kopfmann, S.; Hobeika, E.; Finke, D.; Esser, C.; Diefenbach, A. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 2011, 334, 1561–1565. [Google Scholar] [CrossRef]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Alfonzo-Mendez, M.A.; Alcantara-Hernandez, R.; Garcia-Sainz, J.A. Novel Structural Approaches to Study GPCR Regulation. Int. J. Mol. Sci. 2016, 18, 27. [Google Scholar] [CrossRef] [PubMed]
- Gether, U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr. Rev. 2000, 21, 90–113. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.J.; Deupi, X.; Lebon, G.; Tate, C.G.; Schertler, G.F.; Babu, M.M. Molecular signatures of G-protein-coupled receptors. Nature 2013, 494, 185–194. [Google Scholar] [CrossRef]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef]
- Xiong, Y.; Miyamoto, N.; Shibata, K.; Valasek, M.A.; Motoike, T.; Kedzierski, R.M.; Yanagisawa, M. Short-chain fatty acids stimulate leptin production in adipocytes through the G protein-coupled receptor GPR41. Proc. Natl. Acad. Sci. USA 2004, 101, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Tazoe, H.; Otomo, Y.; Karaki, S.; Kato, I.; Fukami, Y.; Terasaki, M.; Kuwahara, A. Expression of short-chain fatty acid receptor GPR41 in the human colon. Biomed. Res. 2009, 30, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406.e10. [Google Scholar] [CrossRef]
- Wang, A.; Si, H.; Liu, D.; Jiang, H. Butyrate activates the cAMP-protein kinase A-cAMP response element-binding protein signaling pathway in Caco-2 cells. J. Nutr. 2012, 142, 1–6. [Google Scholar] [CrossRef]
- Tazoe, H.; Otomo, Y.; Kaji, I.; Tanaka, R.; Karaki, S.I.; Kuwahara, A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008, 59 (Suppl. 2), 251–262. [Google Scholar]
- Karaki, S.; Mitsui, R.; Hayashi, H.; Kato, I.; Sugiya, H.; Iwanaga, T.; Furness, J.B.; Kuwahara, A. Short-chain fatty acid receptor, GPR43, is expressed by enteroendocrine cells and mucosal mast cells in rat intestine. Cell Tissue Res. 2006, 324, 353–360. [Google Scholar] [CrossRef]
- Karaki, S.; Tazoe, H.; Hayashi, H.; Kashiwabara, H.; Tooyama, K.; Suzuki, Y.; Kuwahara, A. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J. Mol. Histol. 2008, 39, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [Green Version]
- McNelis, J.C.; Lee, Y.S.; Mayoral, R.; van der Kant, R.; Johnson, A.M.; Wollam, J.; Olefsky, J.M. GPR43 Potentiates beta-Cell Function in Obesity. Diabetes 2015, 64, 3203–3217. [Google Scholar] [CrossRef]
- Kim, M.; Friesen, L.; Park, J.; Kim, H.M.; Kim, C.H. Microbial metabolites, short-chain fatty acids, restrain tissue bacterial load, chronic inflammation, and associated cancer in the colon of mice. Eur. J. Immunol. 2018, 48, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Digby, J.E.; Martinez, F.; Jefferson, A.; Ruparelia, N.; Chai, J.; Wamil, M.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arter. Thromb. Vasc. Biol. 2012, 32, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Zandi-Nejad, K.; Takakura, A.; Jurewicz, M.; Chandraker, A.K.; Offermanns, S.; Mount, D.; Abdi, R. The role of HCA2 (GPR109A) in regulating macrophage function. FASEB J. 2013, 27, 4366–4374. [Google Scholar] [CrossRef] [Green Version]
- Gambhir, D.; Ananth, S.; Veeranan-Karmegam, R.; Elangovan, S.; Hester, S.; Jennings, E.; Offermanns, S.; Nussbaum, J.J.; Smith, S.B.; Thangaraju, M.; et al. GPR109A as an anti-inflammatory receptor in retinal pigment epithelial cells and its relevance to diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2208–2217. [Google Scholar] [CrossRef]
- Li, X.; Millar, J.S.; Brownell, N.; Briand, F.; Rader, D.J. Modulation of HDL metabolism by the niacin receptor GPR109A in mouse hepatocytes. Biochem. Pharm. 2010, 80, 1450–1457. [Google Scholar] [CrossRef]
- Irukayama-Tomobe, Y.; Tanaka, H.; Yokomizo, T.; Hashidate-Yoshida, T.; Yanagisawa, M.; Sakurai, T. Aromatic D-amino acids act as chemoattractant factors for human leukocytes through a G protein-coupled receptor, GPR109B. Proc. Natl. Acad. Sci. USA 2009, 106, 3930–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.; Tunaru, S.; Langhans, C.D.; Hanson, J.; Michalski, C.W.; Kolker, S.; Jones, P.M.; Okun, J.G.; Offermanns, S. Deorphanization of GPR109B as a receptor for the beta-oxidation intermediate 3-OH-octanoic acid and its role in the regulation of lipolysis. J. Biol. Chem. 2009, 284, 21928–21933. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Itsubo, C.; Sadakane, K.; Hara, T.; Shinagawa, S.; Koga, H.; Nose, H.; Koshimizu, T.A.; Tsujimoto, G. Production and characterization of a monoclonal antibody against GPR40 (FFAR1; free fatty acid receptor 1). Biochem. Biophys. Res. Commun. 2008, 365, 22–28. [Google Scholar] [CrossRef]
- Cartoni, C.; Yasumatsu, K.; Ohkuri, T.; Shigemura, N.; Yoshida, R.; Godinot, N.; le Coutre, J.; Ninomiya, Y.; Damak, S. Taste preference for fatty acids is mediated by GPR40 and GPR120. J. Neurosci. 2010, 30, 8376–8382. [Google Scholar] [CrossRef]
- Ma, D.; Tao, B.; Warashina, S.; Kotani, S.; Lu, L.; Kaplamadzhiev, D.B.; Mori, Y.; Tonchev, A.B.; Yamashima, T. Expression of free fatty acid receptor GPR40 in the central nervous system of adult monkeys. Neurosci. Res. 2007, 58, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Peat, A.J.; McKeown, S.C.; Corbett, D.F.; Goetz, A.S.; Littleton, T.R.; McCoy, D.C.; Kenakin, T.P.; Andrews, J.L.; Ammala, C.; et al. Pharmacological regulation of insulin secretion in MIN6 cells through the fatty acid receptor GPR40: Identification of agonist and antagonist small molecules. Br. J. Pharm. 2006, 148, 619–628. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, M.; Zhang, S.; Yan, L.; Yang, C.; Lu, W.; Li, Y.; Cheng, H. The role of G protein-coupled receptor 40 in lipoapoptosis in mouse beta-cell line NIT-1. J. Mol. Endocrinol. 2007, 38, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Alquier, T.; Peyot, M.L.; Latour, M.G.; Kebede, M.; Sorensen, C.M.; Gesta, S.; Ronald Kahn, C.; Smith, R.D.; Jetton, T.L.; Metz, T.O.; et al. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes 2009, 58, 2607–2615. [Google Scholar] [CrossRef]
- Latour, M.G.; Alquier, T.; Oseid, E.; Tremblay, C.; Jetton, T.L.; Luo, J.; Lin, D.C.; Poitout, V. GPR40 is necessary but not sufficient for fatty acid stimulation of insulin secretion in vivo. Diabetes 2007, 56, 1087–1094. [Google Scholar] [CrossRef]
- Nagasumi, K.; Esaki, R.; Iwachidow, K.; Yasuhara, Y.; Ogi, K.; Tanaka, H.; Nakata, M.; Yano, T.; Shimakawa, K.; Taketomi, S.; et al. Overexpression of GPR40 in pancreatic beta-cells augments glucose-stimulated insulin secretion and improves glucose tolerance in normal and diabetic mice. Diabetes 2009, 58, 1067–1076. [Google Scholar] [CrossRef]
- Miyamoto, J.; Mizukure, T.; Park, S.B.; Kishino, S.; Kimura, I.; Hirano, K.; Bergamo, P.; Rossi, M.; Suzuki, T.; Arita, M.; et al. A gut microbial metabolite of linoleic acid, 10-hydroxy-cis-12-octadecenoic acid, ameliorates intestinal epithelial barrier impairment partially via GPR40-MEK-ERK pathway. J. Biol. Chem. 2015, 290, 2902–2918. [Google Scholar] [CrossRef]
- Mobraten, K.; Haug, T.M.; Kleiveland, C.R.; Lea, T. Omega-3 and omega-6 PUFAs induce the same GPR120-mediated signalling events, but with different kinetics and intensity in Caco-2 cells. Lipids Health Dis. 2013, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, A.; Tsumaya, K.; Awaji, T.; Katsuma, S.; Adachi, T.; Yamada, M.; Sugimoto, Y.; Miyazaki, S.; Tsujimoto, G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat. Med. 2005, 11, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, C.; Hong, Y.H.; Iga, T.; Hishikawa, D.; Suzuki, Y.; Song, S.H.; Choi, K.C.; Adachi, T.; Hirasawa, A.; Tsujimoto, G.; et al. The regulation of adipogenesis through GPR120. Biochem. Biophys. Res. Commun. 2007, 354, 591–597. [Google Scholar] [CrossRef]
- Ichimura, A.; Hirasawa, A.; Poulain-Godefroy, O.; Bonnefond, A.; Hara, T.; Yengo, L.; Kimura, I.; Leloire, A.; Liu, N.; Iida, K.; et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature 2012, 483, 350–354. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wu, X.; Simonavicius, N.; Tian, H.; Ling, L. Medium-chain fatty acids as ligands for orphan G protein-coupled receptor GPR84. J. Biol. Chem. 2006, 281, 34457–34464. [Google Scholar] [CrossRef]
- Bouchard, C.; Page, J.; Bedard, A.; Tremblay, P.; Vallieres, L. G protein-coupled receptor 84, a microglia-associated protein expressed in neuroinflammatory conditions. Glia 2007, 55, 790–800. [Google Scholar] [CrossRef]
- Venkataraman, C.; Kuo, F. The G-protein coupled receptor, GPR84 regulates IL-4 production by T lymphocytes in response to CD3 crosslinking. Immunol. Lett. 2005, 101, 144–153. [Google Scholar] [CrossRef]
- Imielinski, M.; Baldassano, R.N.; Griffiths, A.; Russell, R.K.; Annese, V.; Dubinsky, M.; Kugathasan, S.; Bradfield, J.P.; Walters, T.D.; Sleiman, P.; et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat. Genet. 2009, 41, 1335–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef]
- Oka, S.; Ota, R.; Shima, M.; Yamashita, A.; Sugiura, T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010, 395, 232–237. [Google Scholar] [CrossRef]
- Zhao, P.; Sharir, H.; Kapur, A.; Cowan, A.; Geller, E.B.; Adler, M.W.; Seltzman, H.H.; Reggio, P.H.; Heynen-Genel, S.; Sauer, M.; et al. Targeting of the orphan receptor GPR35 by pamoic acid: A potent activator of extracellular signal-regulated kinase and beta-arrestin2 with antinociceptive activity. Mol. Pharm. 2010, 78, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Tonai-Kachi, H.; Shinjo, K. Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett. 2006, 580, 5003–5008. [Google Scholar] [CrossRef] [Green Version]
- Deng, H.; Hu, H.; He, M.; Hu, J.; Niu, W.; Ferrie, A.M.; Fang, Y. Discovery of 2-(4-methylfuran-2(5H)-ylidene)malononitrile and thieno[3,2-b]thiophene-2-carboxylic acid derivatives as G protein-coupled receptor 35 (GPR35) agonists. J. Med. Chem. 2011, 54, 7385–7396. [Google Scholar] [CrossRef]
- Burkhardt, A.M.; Maravillas-Montero, J.L.; Carnevale, C.D.; Vilches-Cisneros, N.; Flores, J.P.; Hevezi, P.A.; Zlotnik, A. CXCL17 is a major chemotactic factor for lung macrophages. J. Immunol. 2014, 193, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Maravillas-Montero, J.L.; Burkhardt, A.M.; Hevezi, P.A.; Carnevale, C.D.; Smit, M.J.; Zlotnik, A. Cutting edge: GPR35/CXCR8 is the receptor of the mucosal chemokine CXCL17. J. Immunol. 2015, 194, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Binti Mohd Amir, N.A.S.; Mackenzie, A.E.; Jenkins, L.; Boustani, K.; Hillier, M.C.; Tsuchiya, T.; Milligan, G.; Pease, J.E. Evidence for the Existence of a CXCL17 Receptor Distinct from GPR35. J. Immunol. 2018, 201, 714–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinghaus, D.; Folseraas, T.; Holm, K.; Ellinghaus, E.; Melum, E.; Balschun, T.; Laerdahl, J.K.; Shiryaev, A.; Gotthardt, D.N.; Weismuller, T.J.; et al. Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology 2013, 58, 1074–1083. [Google Scholar] [CrossRef]
- Yang, S.K.; Hong, M.; Choi, H.; Zhao, W.; Jung, Y.; Haritunians, T.; Ye, B.D.; Kim, K.J.; Park, S.H.; Lee, I.; et al. Immunochip analysis identification of 6 additional susceptibility loci for Crohn’s disease in Koreans. Inflamm. Bowel Dis. 2015, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Duboc, H.; Tache, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perino, A.; Pols, T.W.; Nomura, M.; Stein, S.; Pellicciari, R.; Schoonjans, K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J. Clin. Investig. 2014, 124, 5424–5436. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 944. [Google Scholar] [CrossRef]
- Ishii, S.; Kihara, Y.; Shimizu, T. Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J. Biol. Chem. 2005, 280, 9083–9087. [Google Scholar] [CrossRef]
- Ke, X. Presence of multiple independent effects in risk loci of common complex human diseases. Am. J. Hum. Genet. 2012, 91, 185–192. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- Ryder, C.; McColl, K.; Zhong, F.; Distelhorst, C.W. Acidosis promotes Bcl-2 family-mediated evasion of apoptosis: Involvement of acid-sensing G protein-coupled receptor Gpr65 signaling to Mek/Erk. J. Biol. Chem. 2012, 287, 27863–27875. [Google Scholar] [CrossRef] [PubMed]
- Kottyan, L.C.; Collier, A.R.; Cao, K.H.; Niese, K.A.; Hedgebeth, M.; Radu, C.G.; Witte, O.N.; Khurana Hershey, G.K.; Rothenberg, M.E.; Zimmermann, N. Eosinophil viability is increased by acidic pH in a cAMP- and GPR65-dependent manner. Blood 2009, 114, 2774–2782. [Google Scholar] [CrossRef] [Green Version]
- Rosko, A.E.; McColl, K.S.; Zhong, F.; Ryder, C.B.; Chang, M.J.; Sattar, A.; Caimi, P.F.; Hill, B.T.; Al-Harbi, S.; Almasan, A.; et al. Acidosis Sensing Receptor GPR65 Correlates with Anti-Apoptotic Bcl-2 Family Member Expression in CLL Cells: Potential Implications for the CLL Microenvironment. J. Leuk (Los Angel) 2014, 2. [Google Scholar] [CrossRef]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Toma, I.; Kang, J.J.; Sipos, A.; Vargas, S.; Bansal, E.; Hanner, F.; Meer, E.; Peti-Peterdi, J. Succinate receptor GPR91 provides a direct link between high glucose levels and renin release in murine and rabbit kidney. J. Clin. Investig. 2008, 118, 2526–2534. [Google Scholar] [CrossRef] [Green Version]
- Rubic, T.; Lametschwandtner, G.; Jost, S.; Hinteregger, S.; Kund, J.; Carballido-Perrig, N.; Schwarzler, C.; Junt, T.; Voshol, H.; Meingassner, J.G.; et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat. Immunol. 2008, 9, 1261–1269. [Google Scholar] [CrossRef]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical profiling of G protein-coupled receptor expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, W.; Giroux, C.; Cai, Y.; Ekambaram, P.; Dilly, A.K.; Hsu, A.; Zhou, S.; Maddipati, K.R.; Liu, J.; et al. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 2011, 286, 33832–33840. [Google Scholar] [CrossRef]
- Zingoni, A.; Rocchi, M.; Storlazzi, C.T.; Bernardini, G.; Santoni, A.; Napolitano, M. Isolation and chromosomal localization of GPR31, a human gene encoding a putative G protein-coupled receptor. Genomics 1997, 42, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Umemoto, E.; Fujita, S.; Hayashi, A.; Kikuta, J.; Kimura, I.; Haneda, T.; Imai, T.; Inoue, A.; Mimuro, H.; et al. GPR31-dependent dendrite protrusion of intestinal CX3CR1(+) cells by bacterial metabolites. Nature 2019, 566, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.M.; Huang, X.M.; Fan, D.J.; Lin, X.T.; Zhang, F.; Hu, J.C.; Tan, Y.X.; Chen, X.; Zou, Y.F.; Lan, P. Overexpression of G protein-coupled receptor 31 as a poor prognosticator in human colorectal cancer. World J. Gastroenterol. 2018, 24, 4679–4690. [Google Scholar] [CrossRef]
- Smith, K.; McCoy, K.D.; Macpherson, A.J. Use of axenic animals in studying the adaptation of mammals to their commensal intestinal microbiota. Semin. Immunol. 2007, 19, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Niess, J.H.; Leithauser, F.; Adler, G.; Reimann, J. Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J. Immunol. 2008, 180, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, A.H.; Brzezinski, A.; Baker, J.P. Treatment of refractory ulcerative proctosigmoiditis with butyrate enemas. Am. J. Gastroenterol. 1994, 89, 179–183. [Google Scholar]
- Farooq, S.M.; Hou, Y.; Li, H.; O’Meara, M.; Wang, Y.; Li, C.; Wang, J.M. Disruption of GPR35 Exacerbates Dextran Sulfate Sodium-Induced Colitis in Mice. Dig. Dis. Sci. 2018, 63, 2910–2922. [Google Scholar] [CrossRef]
- Tsukahara, T.; Hamouda, N.; Utsumi, D.; Matsumoto, K.; Amagase, K.; Kato, S. G protein-coupled receptor 35 contributes to mucosal repair in mice via migration of colonic epithelial cells. Pharm. Res. 2017, 123, 27–39. [Google Scholar] [CrossRef]
- Lassen, K.G.; McKenzie, C.I.; Mari, M.; Murano, T.; Begun, J.; Baxt, L.A.; Goel, G.; Villablanca, E.J.; Kuo, S.Y.; Huang, H.; et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016, 44, 1392–1405. [Google Scholar] [CrossRef]
- Suzuki, M.; Takaishi, S.; Nagasaki, M.; Onozawa, Y.; Iino, I.; Maeda, H.; Komai, T.; Oda, T. Medium-chain fatty acid-sensing receptor, GPR84, is a proinflammatory receptor. J. Biol. Chem. 2013, 288, 10684–10691. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; Grandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Poritz, L.S.; Garver, K.I.; Green, C.; Fitzpatrick, L.; Ruggiero, F.; Koltun, W.A. Loss of the tight junction protein ZO-1 in dextran sulfate sodium induced colitis. J. Surg. Res. 2007, 140, 12–19. [Google Scholar] [CrossRef]
- Salim, S.Y.; Soderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.X.; Lei, Z.; Wolf, P.G.; Gao, Y.; Guo, Y.M.; Zhang, B.K. Zinc Supplementation, via GPR39, Upregulates PKCzeta to Protect Intestinal Barrier Integrity in Caco-2 Cells Challenged by Salmonella enterica Serovar Typhimurium. J. Nutr. 2017, 147, 1282–1289. [Google Scholar] [CrossRef]
- de Valliere, C.; Vidal, S.; Clay, I.; Jurisic, G.; Tcymbarevich, I.; Lang, S.; Ludwig, M.G.; Okoniewski, M.; Eloranta, J.J.; Kullak-Ublick, G.A.; et al. The pH-sensing receptor OGR1 improves barrier function of epithelial cells and inhibits migration in an acidic environment. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G475–G490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Kabiri, Z.; Greicius, G.; Madan, B.; Biechele, S.; Zhong, Z.; Zaribafzadeh, H.; Edison; Aliyev, J.; Wu, Y.; Bunte, R.; et al. Stroma provides an intestinal stem cell niche in the absence of epithelial Wnts. Development 2014, 141, 2206–2215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Kim, T.Y.; Kim, Y.; Lee, S.H.; Kim, S.; Kang, S.W.; Yang, J.Y.; Baek, I.J.; Sung, Y.H.; Park, Y.Y.; et al. Microbiota-Derived Lactate Accelerates Intestinal Stem-Cell-Mediated Epithelial Development. Cell Host Microbe 2018, 24, 833–846.e6. [Google Scholar] [CrossRef]
- Ho, S.; Pothoulakis, C.; Koon, H.W. Antimicrobial peptides and colitis. Curr. Pharm. Des. 2013, 19, 40–47. [Google Scholar] [PubMed]
- Zhao, Y.; Chen, F.; Wu, W.; Sun, M.; Bilotta, A.J.; Yao, S.; Xiao, Y.; Huang, X.; Eaves-Pyles, T.D.; Golovko, G.; et al. GPR43 mediates microbiota metabolite SCFA regulation of antimicrobial peptide expression in intestinal epithelial cells via activation of mTOR and STAT3. Mucosal. Immunol. 2018, 11, 752–762. [Google Scholar] [CrossRef]
- Sina, C.; Gavrilova, O.; Forster, M.; Till, A.; Derer, S.; Hildebrand, F.; Raabe, B.; Chalaris, A.; Scheller, J.; Rehmann, A.; et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol. 2009, 183, 7514–7522. [Google Scholar] [CrossRef]
- Kamp, M.E.; Shim, R.; Nicholls, A.J.; Oliveira, A.C.; Mason, L.J.; Binge, L.; Mackay, C.R.; Wong, C.H. G Protein-Coupled Receptor 43 Modulates Neutrophil Recruitment during Acute Inflammation. PLoS ONE 2016, 11, e0163750. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Ganji, S.H.; Kashyap, M.L. The mechanism and mitigation of niacin-induced flushing. Int. J. Clin. Pract. 2009, 63, 1369–1377. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Receptor | Ligands | G Protein | Main Expression | Effects on Immune System | Effects on Metabolism | Key IBD-Related Findings |
|---|---|---|---|---|---|---|
| Short chain fat acid | ||||||
| GPR41 (FFAR3) | SCFAs (C2–C6): butyrate, acetate, propionate, valerate | Gαi | Immune cells: peripheral blood monocular cells and macrophages. Enteroendocrine cells, adipocytes, pancreatic islets | DC cells maturation, Th2 development, anti-inflammatory | Regulation of energy balance | Protective effect in DSS and TNBS-induced colitis |
| GPR43 (FFAR2) | SCFAs (C2–C6): butyrate, acetate, propionate, valerate, formate, | Gαi/Gαq | Immune cells: neutrophils, eosinophils. Enteroendocrine cells adipocytes, pancreatic islets | Tumor suppressor, anti-inflammatory | Loss of glucose tolerance, Impaired insulin secretion | Protective effect in DSS and TNBS-induced colitis |
| GPR109A (NACR1) | SCFAs (C4–C8): butyrate and nicotinic acid (niacin) | Gαi/Gαq | Immune cells: DC, neutrophils and macrophages. Intestinal epithelial cells, adipocytes | Inhibition of pro-inflammatory cytokines secretion, anti-inflammatory | Anti-lipolytic | Protective effect in DSS-induced colitis |
| Medium and long-chain fat acid | ||||||
| GPR40 (FFAR1) | C12–C18: medium and long-chain fatty acids | Gαq/11 | Pancreatic cells Enteroendocrine K cells | Anti-inflammatory | Regulation of insulin secretion and glucose tolerance | Protective effect in DSS-induced colitis, Ameliorate intestinal epithelial barrier |
| GPR84 | C9–C14: medium-chain fatty acids | Gαi | Immune cells: leucocytes, neutrophils and macrophages. Adipocytes | Unclear | Unclear | Unstudied |
| GPR120 (FFAR4) | (C12–C22): long-chain fatty acids, unsaturated, ω-3 and ω-6 fatty acids | Gαi/Gαq | Immune cells: macrophages, DC, eosinophils Adipocytes and colon enteroendocrine cells | Inhibition of pro-inflammatory cytokines secretion | Regulation of insulin secretion | Protective effect in IL-10−/− induced chronic colitis, Induce the secretion of antimicrobial peptides |
| Amino acids and related metabolites | ||||||
| GPR35 | kynurenic acid, lysophosphatidic acid and pamoic acid | Gαi | Immune cells: DC, monocytes, neutrophils, macrophages. Intestinal epithelial cells and nervous tissues | Leukocyte recruitment | Unstudied | SNPs associated with IBD, Protective effect in DSS-induced colitis |
| pH-sensitive receptors | ||||||
| GPR65 (TDAG8) | Protons (H+) | Gαs | Immune cells: blood leucocytes. Spleen, thymus, lung and gut | Increase eosinophil viability | Unstudied | SNPs associated with IBD, Protective effect in DSS-induced acute and chronic colitis |
| Succinate | ||||||
| GPR91 (SUCNR1) | Succinate | Gαi/Gαq | Immune cells: DC, macrophages and Platelets. Adipocytes, retinal neurons, liver, heart, intestine and spleen | Migration of Langerhans cells, hematopoiesis, | Hypertensive effects, activation of renin-angiotensin system | Unstudied |
| Bile acid receptors | ||||||
| GPR131 (TGR5) | Lithocholic acid and taurolithocholic acid | Gαs | Immune cells: monocytes and macrophages. Muscle, adipocytes and enteric nervous system | Inflammasome activation, inhibit production of pro-inflammatory cytokine | Insulin resistance | Protective effect in DSS and TNBS-induced colitis. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Melhem, H.; Kaya, B.; Ayata, C.K.; Hruz, P.; Niess, J.H. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells 2019, 8, 450. https://doi.org/10.3390/cells8050450
Melhem H, Kaya B, Ayata CK, Hruz P, Niess JH. Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells. 2019; 8(5):450. https://doi.org/10.3390/cells8050450
Chicago/Turabian StyleMelhem, Hassan, Berna Kaya, C. Korcan Ayata, Petr Hruz, and Jan Hendrik Niess. 2019. "Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease" Cells 8, no. 5: 450. https://doi.org/10.3390/cells8050450
APA StyleMelhem, H., Kaya, B., Ayata, C. K., Hruz, P., & Niess, J. H. (2019). Metabolite-Sensing G Protein-Coupled Receptors Connect the Diet-Microbiota-Metabolites Axis to Inflammatory Bowel Disease. Cells, 8(5), 450. https://doi.org/10.3390/cells8050450