Functionally Significant Features in the 5′ Untranslated Region of the ABCA1 Gene and Their Comparison in Vertebrates



Abstract
:
1. Introduction
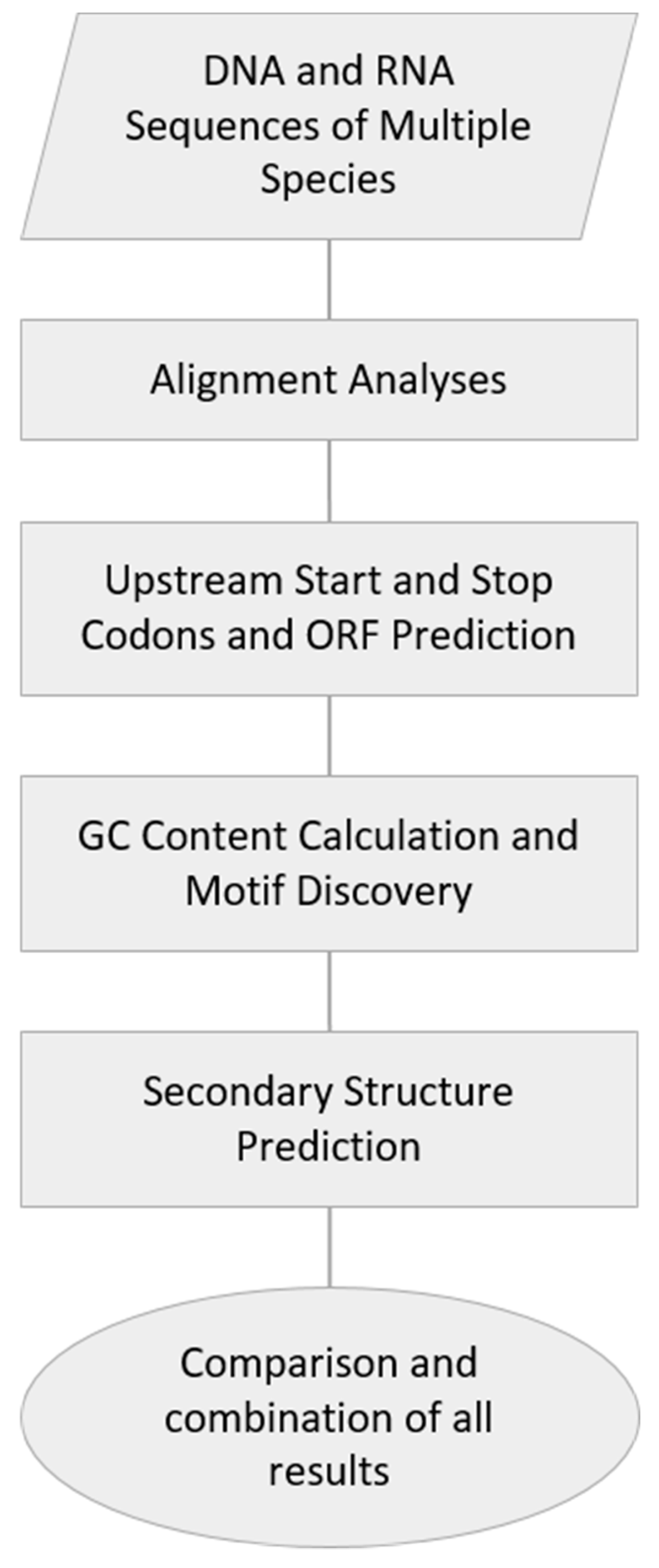
2. Materials and Methods
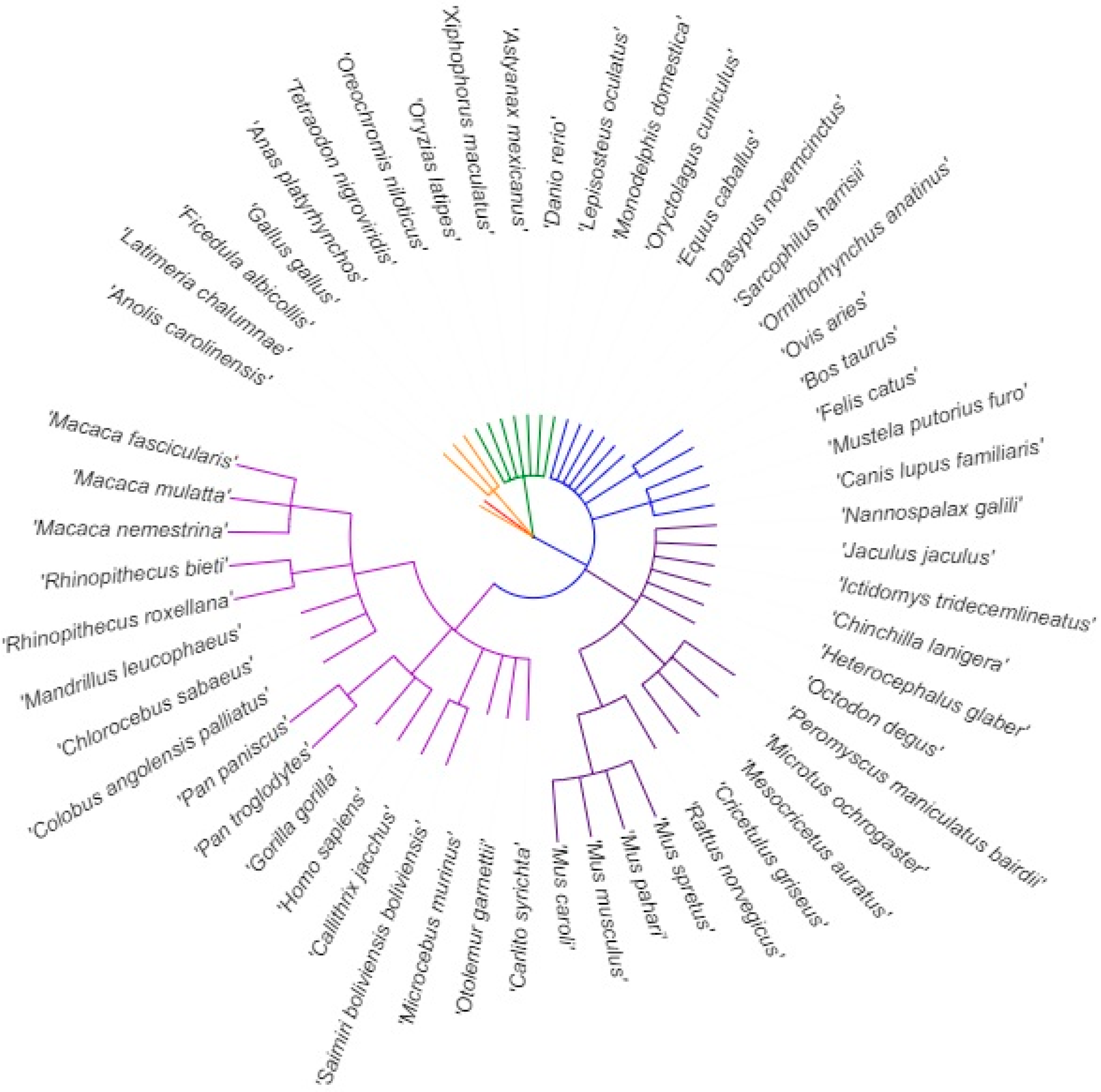
2.1. DNA and RNA Sequences of Multiple Species
2.2. Alignment Analyses
2.3. Upstream Start and Stop Codons and ORF Prediction
2.4. GC Content Calculation and Motif Discovery
2.5. Secondary Structure Prediction
2.6. Statistics
3. Results
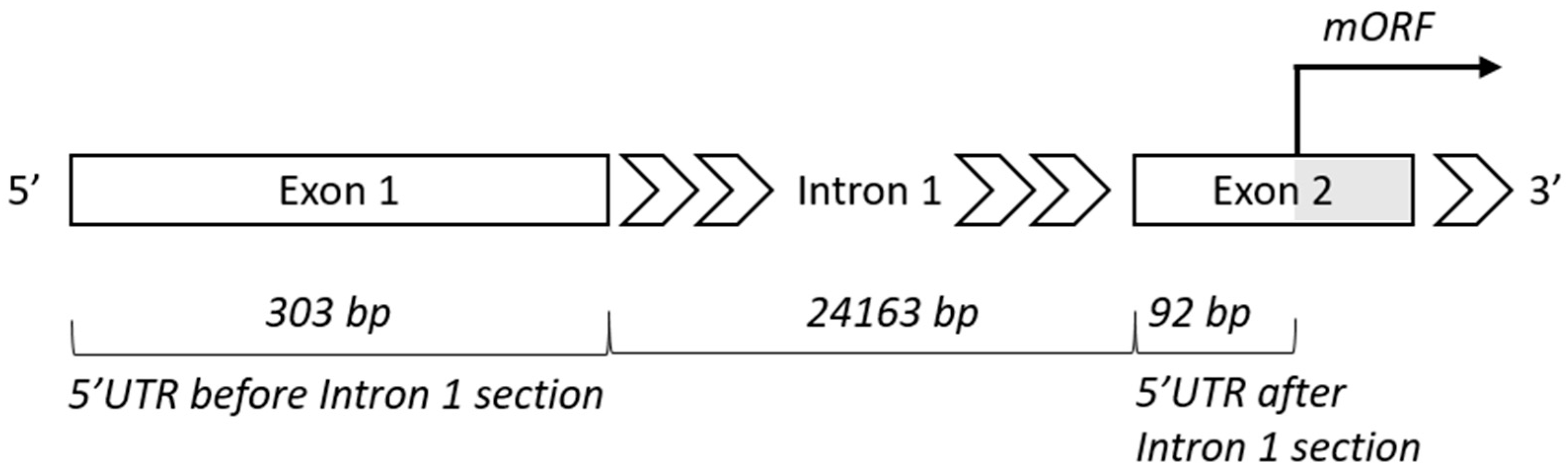
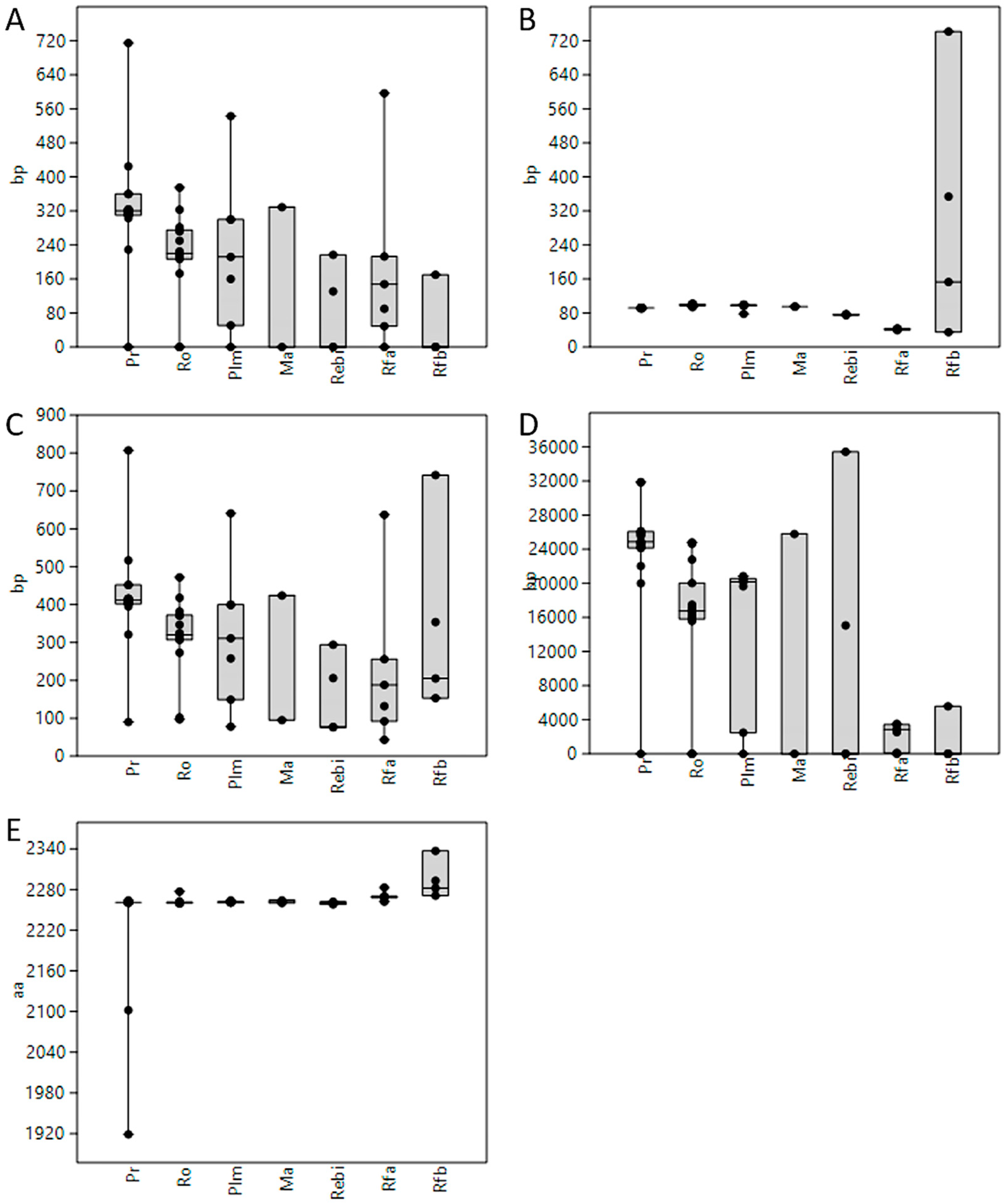
3.1. Length of 5′UTR Sections Located after-Intron-1 are Conserved within Main Vertebrate Subgroups
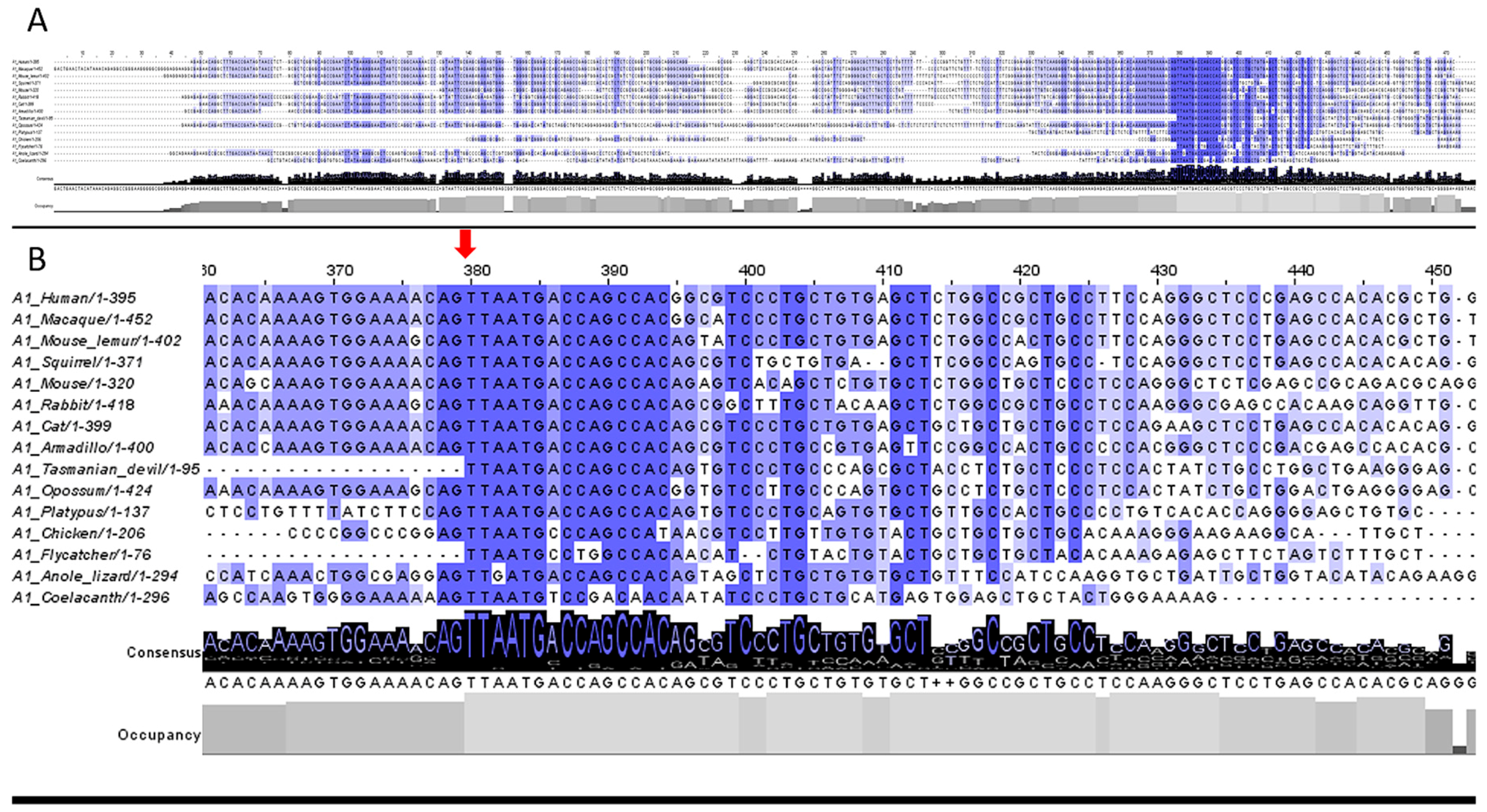
3.2. 5′UTR Sections Located after-Intron-1 Start with Highly Conserved Sequences
3.3. Upstream ATG is Localized within the Highly Conserved Sequence at the Start of the 5′UTR after-Intron-1 Section
3.4. Upstream ORF Starting with Highly Conserved Upstream ATG Expanded during Vertebrate Evolution and Became Overlapping with Main ORF
3.5. GC Content Showed Great Variability among 5′UTRs of Extant Vertebrates with Placental Mammals Having the Highest Percentage
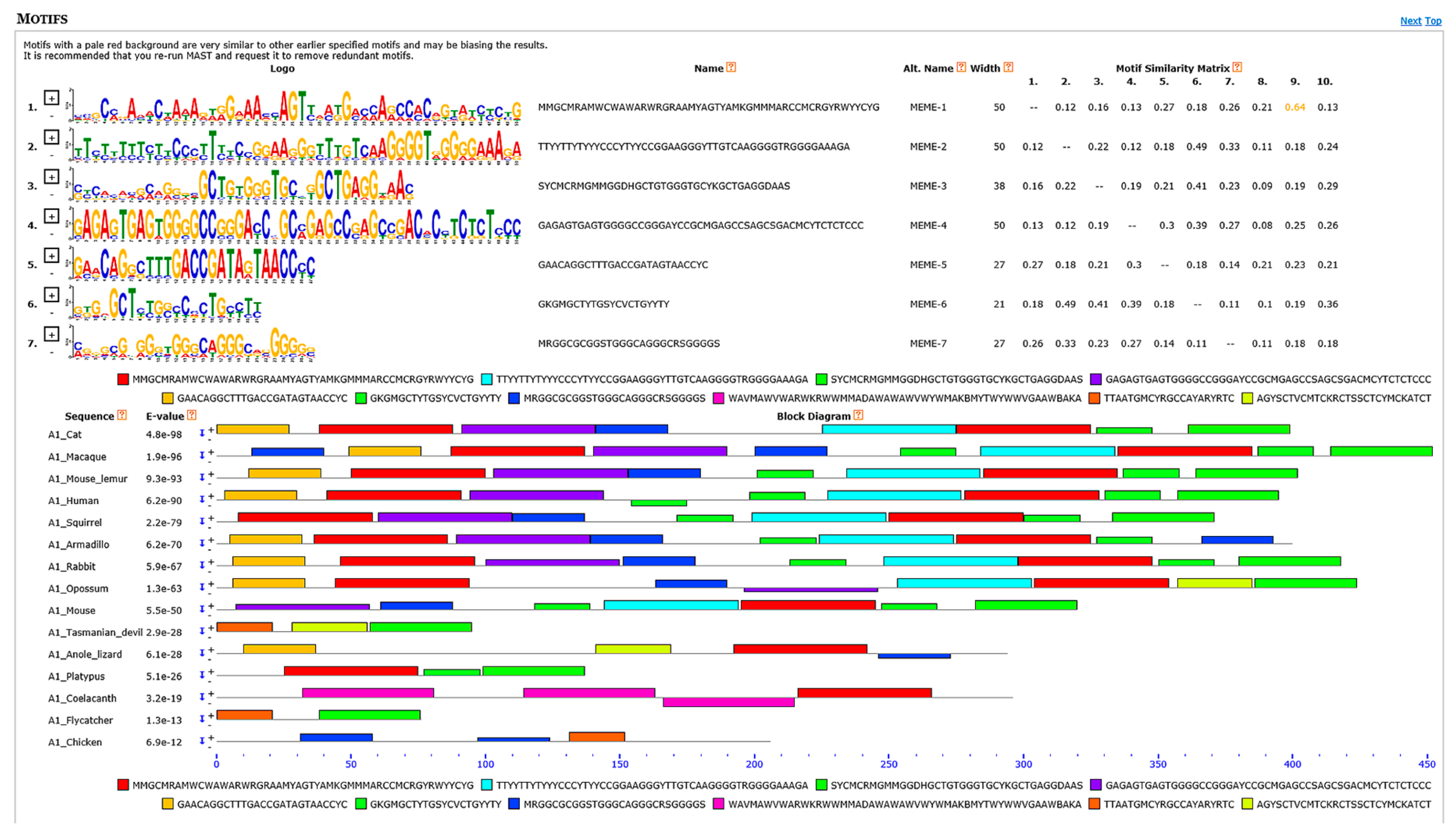
3.6. Motif Discovery Analyses Showed Several Highly Conserved Elements Mainly among Transcriptional Regulatory Motifs and Intron Splicing Enhancers
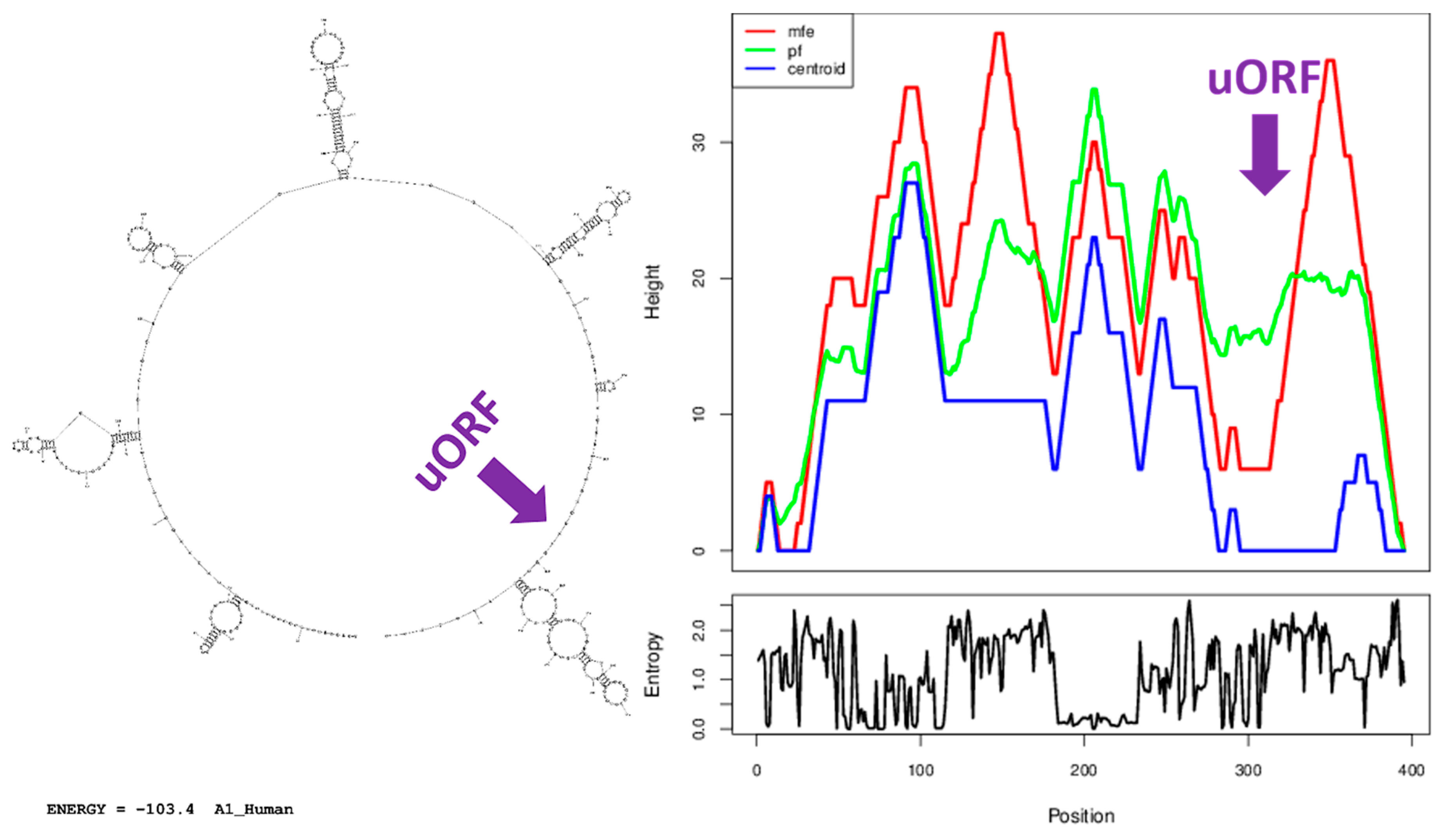
3.7. A Similar Number of Hairpin Loops and Their Distribution in 5′UTRs is Present throughout Extant Vertebrates
3.8. Analysis of 5′UTR Variants Annotated in Ensembl Database
4. Discussion
4.1. 5′UTR Length
4.2. Intron 1—Length and Position
4.3. Upstream ORF—Position, Function, and Evolution
4.4. GC Content and Motifs
4.5. Secondary Structure
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5′UTR | 5′ untranslated region |
| ESE | exon splicing enhancer |
| ESS | exon splicing silencer |
| HDL | high density lipoprotein |
| ISE | intron splicing enhancer |
| mORF | main open reading frame |
| nts | nucleotides |
| SNP | single nucleotide polymorphism |
| sPEP | small peptide |
| TIS | translation initiation site |
| TSS | transcription start site |
| uATG | upstream ATG codon |
| uORF | upstream open reading frame |
References
- Guo, Y.; Long, J.; He, J.; Li, C.; Cai, Q.; Shu, X.; Zheng, W.; Li, C. Exome sequencing generates high quality data in non-target regions. BMC Genom. 2012, 13, 194. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001, 11, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.; Singh, A.; Keelan, J. Transport of lipids by ABC proteins: Interactions and implications for cellular toxicity, viability and function. Chem. Biol. Interact. 2009, 180, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Nürenberg, E.; Tampé, R. Tying up loose ends: Ribosome recycling in eukaryotes and archaea. Trends Biochem. Sci. 2013, 38, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Iida, A.; Saito, S.; Sekine, A.; Kitamura, Y.; Kondo, K.; Mishima, C.; Osawa, S.; Harigae, S.; Nakamura, Y. High-density single-nucleotide polymorphism (SNP) map of the 150-kb region corresponding to the human ATP-binding cassette transporter A1 (ABCA1) gene. J. Hum. 2001, 46, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M. Is ABCA1 a lipid transfer protein? J. Lipid Res. 2018, 59, 749–763. [Google Scholar] [CrossRef] [Green Version]
- Araujo, P.; Yoon, K.; Ko, D.; Smith, A.; Qiao, M.; Suresh, U.; Burns, S.; Penalva, L. Before it gets started: Regulating translation at the 5′ UTR. Comp. Func. Genom. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haimov, O.; Sinvani, H.; Dikstein, R. Cap-dependent, scanning-free translation initiation mechanisms. BBA-Gene Regul. Mech. 2015, 1849, 1313–1318. [Google Scholar] [CrossRef]
- Jackson, R.; Hellen, C.; Pestova, T. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Rojas-Duran, M.; Gilbert, W. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 2012, 18, 2299–2305. [Google Scholar] [CrossRef] [Green Version]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2017, 19, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Pezeshkpoor, B.; Berkemeier, A.; Czogalla, K.; Oldenburg, J.; El-Maarri, O. Evidence of pathogenicity of a mutation in 3′ untranslated region causing mild haemophilia A. Haemophilia 2016, 22, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Piraino, S.; Furney, S. Beyond the exome: The role of non-coding somatic mutations in cancer. Ann. Oncol. 2015, 27, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Cenik, C.; Derti, A.; Mellor, J.; Berriz, G.; Roth, F. Genome-wide functional analysis of human 5′ untranslated region introns. Genome Biol. 2010, 11, R29. [Google Scholar] [CrossRef] [PubMed]
- Bicknell, A.; Cenik, C.; Chua, H.; Roth, F.; Moore, M. Introns in UTRs: Why we should stop ignoring them? Bioessays 2012, 34, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Jo, B.; Choi, S. Introns: The functional benefits of introns in genomes. Genom. Inform. 2015, 13, 112. [Google Scholar] [CrossRef]
- Aken, B.; Achuthan, P.; Akanni, W.; Amode, M.; Bernsdorff, F.; Bhai, J.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; et al. Ensembl 2017. Nucleic Acids Res. 2016, 45, D635–D642. [Google Scholar] [CrossRef]
- Rodriguez, J.; Rodriguez-Rivas, J.; Di Domenico, T.; Vázquez, J.; Valencia, A.; Tress, M. APPRIS 2017: Principal isoforms for multiple gene sets. Nucleic Acids Res. 2017, 46, D213–D217. [Google Scholar] [CrossRef]
- Vilella, A.; Severin, J.; Ureta-Vidal, A.; Heng, L.; Durbin, R.; Birney, E. EnsemblCompara GeneTrees: Complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2008, 19, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Haubold, B.; Wiehe, T. Comparative genomics: Methods and applications. Naturwissenschaften 2004, 91, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Procter, J.; Martin, D.; Clamp, M.; Barton, G. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.; Sugnet, C.; Furey, T.; Roskin, K.; Pringle, T.; Zahler, A.; Haussler, A. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Skarshewski, A.; Stanton-Cook, M.; Huber, T.; Al Mansoori, S.; Smith, R.; Beatson, S.; Rothnagel, J. uPEPperoni: An online tool for upstream open reading frame location and analysis of transcript conservation. BMC Bioinform. 2014, 15, 36. [Google Scholar] [CrossRef] [PubMed]
- Tikole, S.; Sankararamakrishnan, R. Prediction of translation initiation sites in human mRNA sequences with AUG start codon in weak Kozak context: A neural network approach. Biochem. Biophys. Res. Commun. 2008, 369, 1166–1168. [Google Scholar] [CrossRef] [PubMed]
- Grillo, G.; Turi, A.; Licciulli, F.; Mignone, F.; Liuni, S.; Banfi, S.; Gennarino, V.; Horner, D.; Pavesi, G.; Picardi, E.; et al. UTRdb and UTRsite (RELEASE 2010): A collection of sequences and regulatory motifs of the untranslated regions of eukaryotic mRNAs. Nucleic Acids Res. 2010, 38, D75–D80. [Google Scholar] [CrossRef]
- Chang, T.; Huang, H.; Hsu, J.; Weng, S.; Horng, J.; Huang, H. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinform. 2013, 14, S4. [Google Scholar]
- Bailey, T.; Bodén, M.; Buske, F.; Frith, M.; Grant, Ch.; Clementi, L.; Ren, J.; Li, W.; Noble, W. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Pesole, G.; Grillo, G.; Larizza, A.; Liuni, S. The untranslated regions of eukaryotic mRNAs: Structure, function, evolution and bioinformatic tools for their analysis. Brief. Bioinform. 2000, 1, 236–249. [Google Scholar] [CrossRef] [Green Version]
- Pesole, G.; Mignone, F.; Gissi, C.; Grillo, G.; Licciulli, F.; Liuni, S. Structural and functional features of eukaryotic mRNA untranslated regions. Gene 2001, 276, 73–81. [Google Scholar] [CrossRef]
- Mazumder, B.; Seshadri, V.; Fox, P. Translational control by the 3′-UTR: The ends specify the means. Trends Biochem. Sci. 2003, 28, 91–98. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J. The origins of genome complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Scofield, D.; Hong, X. The evolution of transcription-initiation sites. Mol. Biol. Evol. 2005, 22, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Barton, N. Genome size: Does bigger mean worse? Curr. Biol. 2004, 14, R233–R235. [Google Scholar] [CrossRef] [PubMed]
- Daubin, V.; Moran, N. Comment on “The origins of genome complexity”. Science 2004, 306, 978. [Google Scholar] [CrossRef] [PubMed]
- Reuter, M.; Engelstadter, J.; Fontanillas, P.; Hurst, L. A Test of the null model for 5′ UTR evolution based on GC content. Mol. Biol. Evol. 2008, 25, 801–804. [Google Scholar] [CrossRef]
- Vinogradov, A.; Anatskaya, O. Organismal complexity, cell differentiation and gene expression: Human over mouse. Nucleic Acids Res. 2007, 35, 6350–6356. [Google Scholar] [CrossRef]
- Whitney, K.; Garland, T. Did genetic drift drive increases in genome complexity? PLoS Genet. 2010, 6, e1001080. [Google Scholar] [CrossRef]
- Chen, C.; Lin, H.; Pan, C.; Chen, F. The plausible reason why the length of 5′ untranslated region is unrelated to organismal complexity. BMC Res. Notes 2011, 4, 312. [Google Scholar] [CrossRef]
- Chen, C.; Lin, H.; Pan, C.; Chen, F. The genomic features that affect the lengths of 5′ untranslated regions in multicellular eukaryotes. BMC Bioinform. 2011, 12, S3. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Wardell, S.; Kleffmann, T.; Brown, C. The exon-intron gene structure upstream of the initiation codon predicts translation efficiency. Nucleic Acids Res. 2018, 46, 4575–4591. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Scofield, D.; Lynch, M. Intron size, abundance, and distribution within untranslated regions of genes. Mol. Biol. Evol. 2006, 23, 2392–2404. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, M.; Long, M. Intron—Exon structures of eukaryotic model organisms. Nucleic Acids Res. 1999, 27, 3219–3228. [Google Scholar] [PubMed]
- Vinogradov, A. Intron—Genome size relationship on a large evolutionary scale. J. Mol. Evol. 1999, 49, 376–384. [Google Scholar] [CrossRef]
- Larizza, A.; Makalowski, W.; Pesole, G.; Saccone, C. Evolutionary dynamics of mammalian mRNA untranslated regions by comparative analysis of orthologous human, artiodactyl and rodent gene pairs. Comput. Chem. 2002, 26, 479–490. [Google Scholar] [CrossRef]
- Shabalina, S.; Ogurtsov, A.; Rogozin, I.; Koonin, E.; Lipman, D. Comparative analysis of orthologous eukaryotic mRNAs: Potential hidden functional signals. Nucleic Acids Res. 2004, 32, 1774–1782. [Google Scholar] [CrossRef]
- Vinogradov, A. “Genome design” model: Evidence from conserved intronic sequence in human-mouse comparison. Genome Res. 2006, 16, 347–354. [Google Scholar] [CrossRef]
- Pozzoli, U.; Menozzi, G.; Comi, G.; Cagliani, R.; Bresolin, N.; Sironi, M. Intron size in mammals: Complexity comes to terms with economy. Trends Genet. 2007, 23, 20–24. [Google Scholar] [CrossRef]
- LaConte, L.; Mukherjee, K. Structural constraints and functional divergences in CASK evolution. Biochem. Soc. Trans. 2013, 41, 1017–1022. [Google Scholar] [CrossRef] [Green Version]
- Kozak, M. Pushing the limits of the scanning mechanism for initiation of translation. Gene 2002, 299, 1–34. [Google Scholar] [CrossRef]
- Rogozin, I.; Kochetov, A.; Kondrashov, F.; Koonin, E.; Milanesi, L. Presence of ATG triplets in 5′ untranslated regions of eukaryotic cDNAs correlates with a weak’ context of the start codon. Bioinformatics 2001, 17, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Iacono, M.; Mignone, F.; Pesole, G. uAUG and uORFs in human and rodent 5′untranslated mRNAs. Gene 2005, 349, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.; Pagliarini, D.; Mootha, V. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 2009, 106, 7507–7512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churbanov, A.; Rogozin, I.; Babenko, V.; Ali, H.; Koonin, E. Evolutionary conservation suggests a regulatory function of AUG triplets in 5′-UTRs of eukaryotic genes. Nucleic Acids Res. 2005, 33, 5512–5520. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Yachie, N.; Okada, Y.; Saito, R.; Tomita, M. Bioinformatic analysis of post-transcriptional regulation by uORF in human and mouse. FEBS Lett. 2007, 581, 4184–4188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Song, W. Leaky scanning and reinitiation regulate BACE1 gene expression. Mol. Cell Biol. 2006, 26, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Kochetov, A. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays 2008, 30, 683–691. [Google Scholar] [CrossRef]
- Kozak, M. Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol. Cell Biol. 1987, 7, 3438–3445. [Google Scholar] [CrossRef]
- Luukkonen, B.; Tan, W.; Schwartz, S. Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mRNAs is determined by the length of the upstream open reading frame and by intercistronic distance. J. Virol. 1995, 69, 4086–4094. [Google Scholar]
- Kochetov, A.; Ahmad, S.; Ivanisenko, V.; Volkova, O.; Kolchanov, N.; Sarai, A. uORFs, reinitiation and alternative translation start sites in human mRNAs. FEBS Lett. 2008, 582, 1293–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wethmar, K.; Smink, J.; Leutz, A. Upstream open reading frames: Molecular switches in (patho)physiology. Bioessays 2010, 32, 885–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, M.; Chen, F. Selective constraint on the upstream open reading frames that overlap with coding sequences in animals. PLoS ONE 2012, 7, e48413. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, C.; Peixeiro, I.; Romão, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013, 9, e1003529. [Google Scholar] [CrossRef] [PubMed]
- Somers, J.; Pöyry, T.; Willis, A. A perspective on mammalian upstream open reading frame function. Int. J. Biochem. Cell Biol. 2013, 45, 1690–1700. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S.; Rothnagel, J. Emerging evidence for functional peptides encoded by short open reading frames. Nat. Rev. Genet. 2014, 15, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.; Levesque, S.; Hunting, D.; Cohen, A.; Roucou, X. Recognition of the polycistronic nature of human genes is critical to understanding the genotype-phenotype relationship. Genome Res. 2018, 28, 609–624. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G.; Olofsson, B.; Filipski, J.; Zerial, M.; Salinas, J.; Cuny, G.; Meunier-Rotival, M.; Rodier, F. The mosaic genome of warm-blooded vertebrates. Science 1985, 228, 953–958. [Google Scholar] [CrossRef]
- Romiguier, J.; Ranwez, V.; Douzery, E.; Galtier, N. Contrasting GC-content dynamics across 33 mammalian genomes: Relationship with life-history traits and chromosome sizes. Genome Res. 2010, 20, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Duret, L.; Semon, M.; Piganeau, G.; Mouchiroud, D.; Galtier, N. Vanishing GC-rich isochores in mammalian genomes. Genetics 2002, 162, 1837–1847. [Google Scholar]
- Gu, J.; Li, W. Are GC-rich isochores vanishing in mammals? Gene 2006, 385, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Sari-Ak, D.; Haffke, M.; Trowitzsch, S.; Berger, I. Zooming in on transcription preinitiation. J. Mol. Biol. 2016, 428, 2581–2591. [Google Scholar] [CrossRef] [PubMed]
- Mognol, G.; Carneiro, F.; Robbs, B.; Faget, D.; Viola, J. Cell cycle and apoptosis regulation by NFAT transcription factors: New roles for an old player. Cell Death Dis. 2016, 7, e2199. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Shu, F.; Chen, Y.; Chen, Y.; Lan, Y.; Duan, X.; Zhao, S.; Zeng, G. The prognostic value of HOXA13 in solid tumors: A meta-analysis. Clin. Chim. Acta 2018, 483, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Xiong, T.; Xu, G.; Huang, X.; Lu, K.; Xie, W.; Yin, K.; Tu, J. ATP-binding cassette transporter A1: A promising therapy target for prostate cancer. Mol. Clin. Oncol. 2018, 8, 9–14. [Google Scholar] [CrossRef]
- Kozak, M. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc. Natl. Acad. Sci. USA 1990, 87, 8301–8305. [Google Scholar] [CrossRef]
- Kochetov, A.; Palyanov, A.; Titov, I.; Grigorovich, D.; Sarai, A.; Kolchanov, N. AUG_hairpin: Prediction of a downstream secondary structure influencing the recognition of a translation start site. BMC Bioinform. 2007, 8, 318. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Ogurtsov, A.; Shabalina, S.; Koonin, E. Adaptation of mRNA structure to control protein folding. RNA Biol. 2017, 14, 1649–1654. [Google Scholar] [CrossRef]
- Ringner, M.; Krogh, M. Folding free energies of 5′-UTRs impact post-transcriptional regulation on a genomic scale in yeast. PLoS Comput. Biol. 2005, 1, e72. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | uORF | mORF | ||||
|---|---|---|---|---|---|---|
| Start | Stop | Length | Protein Sequence | Start | ||
| nts | aa | |||||
| Human | 307 | 417 | 111 | 36 | MTSHGVPAVSSGRCLPGLPSHTLGVLAEGTWLVGLS | 396 |
| Macaque | 364 | 474 | 111 | 36 | MTSHGIPAVSSGRCLPGLLSHTLWVLAEGTWLVGLS | 453 |
| Mouse lemur | 314 | 424 | 111 | 36 | MTSHSIPAVSSGHCLPGLLSHTLWVPAEVTWLVGPS | 403 |
| Rabbit | 327 | 440 | 114 | 37 | MTSHSGFATSSGRCLQGRATSRLPWVPAEVTWPAGLS | 419 |
| Mouse | 224 | 319 | 96 | 31 | MTSHRVTALCSGCSLQGSRAADAGRCGCRLW | 321 |
| Squirrel | 279 | 365 | 87 | 28 | MTSHSVCCELRPVPPGLLSHTQVALGAG | 372 |
| Cat | 304 | 393 | 90 | 29 | MTSHSVPAVSCCCCLQKLLSHTQVAAAAG | 400 |
| Armadillo | 304 | 399 | 96 | 31 | MTSHSVPAVSSGHCPHGLPTSHTQVAWARLR | 401 |
| Tasmanian devil | 4 | 66 | 63 | 20 | MTSHSVPAQRYLCSLHYLPG | 96 |
| Opossum | 333 | 395 | 63 | 20 | MTSHGVLAQCCLCSLHYLLD | 425 |
| Platypus | 54 | 131 | 78 | 25 | MTSHSVPAVCCCHCPCHTRGAVPAC | 138 |
| Chicken | 135 | 200 | 66 | 21 | MPSHNVLVVYCCCCTKGRRHC | 207 |
| Flycatcher | 4 | 60 | 57 | 18 | MPGHNICTVLLLLHKESF | 77 |
| Anole lizard | 221 | 271 | 51 | 16 | MTSHSSSAVCCFHPRC | 295 |
| Coelacanth | 245 | 274 | 30 | 9 | MSDNNIPAA | 297 |
| Species | uORF nt Context | mORF nt Context |
|---|---|---|
| Human | AAACAGTTAATGACCAGCCAC | TGAGGGAACATGGCTTGTTGG |
| Macaque | AAACAGTTAATGACCAGCCAC | TGAGGGAACATGGCTTGTTGG |
| Mouse lemur | AAGCAGTTAATGACCAGCCAC | TGAGGTGACATGGCTTGTTGG |
| Rabbit | AAGCAGTTAATGACCAGCCAC | TGAGGTAACATGGCCTGCTGG |
| Mouse | AAACAGTTAATGACCAGCCAC | TGTGGTGACATGGCTTGTTGG |
| Squirrel | AAACAGTTAATGACCAGCCAC | TGAGGTAACATGGCTTATTGG |
| Cat | AAACAGTTAATGACCAGCCAC | TGAGGAAACATGGCTTACTGG |
| Armadillo | AAACAGTTAATGACCAGCCAC | TGAGGTAACATGGCTTGCTGG |
| Tasmanian devil | TTAATGACCAGCCAC | TGAGGAAAGATGGCTTTTTGG |
| Opossum | AAGCAGTTAATGACCAGCCAC | TGAGGAGAGATGGCCTTTTGG |
| Platypus | TTCCAGTTAATGACCAGCCAC | TGAGGAAAGATGGCTTTTTGG |
| Chicken | CCGGAGTTAATGCCCAGCCAT | TGAAGAACGATGGCATTTTGG |
| Flycatcher | TTAATGCCTGGCCAC | TGAAGGAAGATGGCTTTCTGG |
| Anole lizard | GAGGAGTTGATGACCAGCCAC | AGAAGGAAGATGGCCTTCTGG |
| Coelacanth | AAAAAGTTAATGTCCGACAAC | TGGGAAAAGATGACTTTCTGG |
| Species | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Motif Type | Motif Name | Hu. | Ma. | Mo. le. | Sq. | Mo. | Ra. | Cat | Ar. | Ta. de. | Op. | Pl. | Chi. | Fl. | An. li. | Co. | Nr. of Hits |
| ESE | beta-globin ex. 2 | • | • | 2 | |||||||||||||
| ct/cgrp | • | • | • | 3 | |||||||||||||
| sc35 | • | • | • | • | • | • | • | • | • | 9 | |||||||
| ESS | fibronectin eda ex. | • | • | • | • | • | • | • | 7 | ||||||||
| ISE | cftr, in. 9 | • | • | • | 3 | ||||||||||||
| ighg2 cgamma2 in. 1 | • | • | • | • | • | • | • | 7 | |||||||||
| ctnt, ex. 5 | • | • | • | • | • | • | • | • | 8 | ||||||||
| gh-1 in. 3 | • | • | • | • | • | • | • | • | • | • | 10 | ||||||
| rho-ind. ter. | Rho-ind. ter. | • | • | • | • | 4 | |||||||||||
| TRM | Sp1 | • | • | 2 | |||||||||||||
| Msx-1 | • | • | 2 | ||||||||||||||
| Freac | • | • | 2 | ||||||||||||||
| c-Ets-1_p54 | • | • | 2 | ||||||||||||||
| SMAD | • | • | 2 | ||||||||||||||
| GATA-5 | • | • | 2 | ||||||||||||||
| ZNF333 | • | • | 2 | ||||||||||||||
| ELF1 | • | • | • | 3 | |||||||||||||
| SPI1 | • | • | • | 3 | |||||||||||||
| MZF1 | • | • | • | 3 | |||||||||||||
| MYB | • | • | • | 3 | |||||||||||||
| AP-2 | • | • | • | 3 | |||||||||||||
| BEN | • | • | • | 3 | |||||||||||||
| Kid3 | • | • | • | • | 4 | ||||||||||||
| PEA3 | • | • | • | • | 4 | ||||||||||||
| E2F-3 | • | • | • | • | 4 | ||||||||||||
| GABP | • | • | • | • | 4 | ||||||||||||
| ZF5 | • | • | • | • | 4 | ||||||||||||
| SOX10 | • | • | • | • | 4 | ||||||||||||
| Elk-1 | • | • | • | • | • | 5 | |||||||||||
| ER81 | • | • | • | • | • | 5 | |||||||||||
| ETV7 | • | • | • | • | • | 5 | |||||||||||
| LXR_direct | • | • | • | • | • | 5 | |||||||||||
| TFIIA | • | • | • | • | • | • | • | 7 | |||||||||
| TATA | • | • | • | • | • | • | • | • | 8 | ||||||||
| NFAT1 | • | • | • | • | • | • | • | • | 8 | ||||||||
| NF-AT4 | • | • | • | • | • | • | • | • | 8 | ||||||||
| HOXA13 | • | • | • | • | • | • | • | • | 8 | ||||||||
| UTR motifs | Musashi bin. El. (MBE) | • | • | • | • | • | • | 6 | |||||||||
| microRNA target sites | hsa-miR-5581-5p | • | • | 2 | |||||||||||||
| hsa-miR-3194-3p | • | • | 2 | ||||||||||||||
| hsa-miR-4435 | • | • | • | 3 | |||||||||||||
| hsa-miR-4474-3p | • | • | • | • | • | • | • | • | • | 9 | |||||||
| Species | No. of Hairpin Loops | |||
|---|---|---|---|---|
| RNAstructure | RNAfold | |||
| Before In. 1 Section | After In. 1 Section | Before In. 1 Section | After In. 1 Section | |
| Human | 6 | 1 | 6 | 1 |
| Mouse lemur | 4 | 1 | 5 | 2 |
| Rabbit | 7 | 1 | 7 | 1 |
| Mouse | 5 | 1 | 4 | 2 |
| Armadillo | 7 | 1 | 5 | 1 |
| Opossum | 7 | 1 | 5 | 1 |
| Platypus | 2 | 2 | ||
| Flycatcher | 2 | 2 | ||
| Anole lizard | 7 | 1 | 6 | 1 |
| Coelacanth | 6 | 1 | 4 | 1 |
| DNA and RNA Sequences of Multiple Species | |
| Ensembl, UCSC | genome browsers for the retrieval of genomic information |
| Taxonomy Browser (NCBI) | builds trees based on the classification in the NCBI taxonomy database |
| iTOL | phylogenetic tree development and visualisation |
| EMBOSS Transeq | translates nucleic acid sequences to their corresponding peptide sequences |
| Transcription and Translation Tool (ExPASy) | transcription, translation, reverse transcription |
| Alignment analyses | |
| Jalview | multiple sequence alignment editing, visualisation and analysis |
| EMBL-EBI, UCSC, NCBI | bioinformatics resources including alignment programs |
| Upstream start and stop codons and ORF prediction | |
| ORF Finder | searches for open reading frames in DNA sequences |
| NetStart | produces neural network predictions of translation start in nucleotide sequences |
| uPEPperoni | location and identification of upstream open reading frames that have the potential to encode bioactive peptides |
| WeakAUG | predict the initiation site of mRNA sequences that lack the preferred nucleotides |
| GC content calculation and motif discovery | |
| GC Content Calculator | GC content, GC Content Distribution Plots |
| UTRscan | finds motifs that characterize 3′UTR and 5′UTR sequences |
| RegRNA | identifies functional RNA motifs and sites in RNA sequences |
| MEME | discovers novel motifs in nucleotide or protein sequences |
| Secondary structure prediction | |
| RNAstructure, RNAfold | predict RNA secondary structures |
| Statistics | |
| PAST | scientific data analysis, statistics |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvorak, P.; Leupen, S.; Soucek, P. Functionally Significant Features in the 5′ Untranslated Region of the ABCA1 Gene and Their Comparison in Vertebrates. Cells 2019, 8, 623. https://doi.org/10.3390/cells8060623
Dvorak P, Leupen S, Soucek P. Functionally Significant Features in the 5′ Untranslated Region of the ABCA1 Gene and Their Comparison in Vertebrates. Cells. 2019; 8(6):623. https://doi.org/10.3390/cells8060623
Chicago/Turabian StyleDvorak, Pavel, Sarah Leupen, and Pavel Soucek. 2019. "Functionally Significant Features in the 5′ Untranslated Region of the ABCA1 Gene and Their Comparison in Vertebrates" Cells 8, no. 6: 623. https://doi.org/10.3390/cells8060623
APA StyleDvorak, P., Leupen, S., & Soucek, P. (2019). Functionally Significant Features in the 5′ Untranslated Region of the ABCA1 Gene and Their Comparison in Vertebrates. Cells, 8(6), 623. https://doi.org/10.3390/cells8060623