Identifying microRNAs and Their Editing Sites in Macaca mulatta

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Collection of Tissue Samples and sRNA-Sequencing
2.2. Identifying Conserved miRNAs of Macaca mulatta
2.3. Expression Patterns of Conserved miRNAs in Different Tissues/Organs
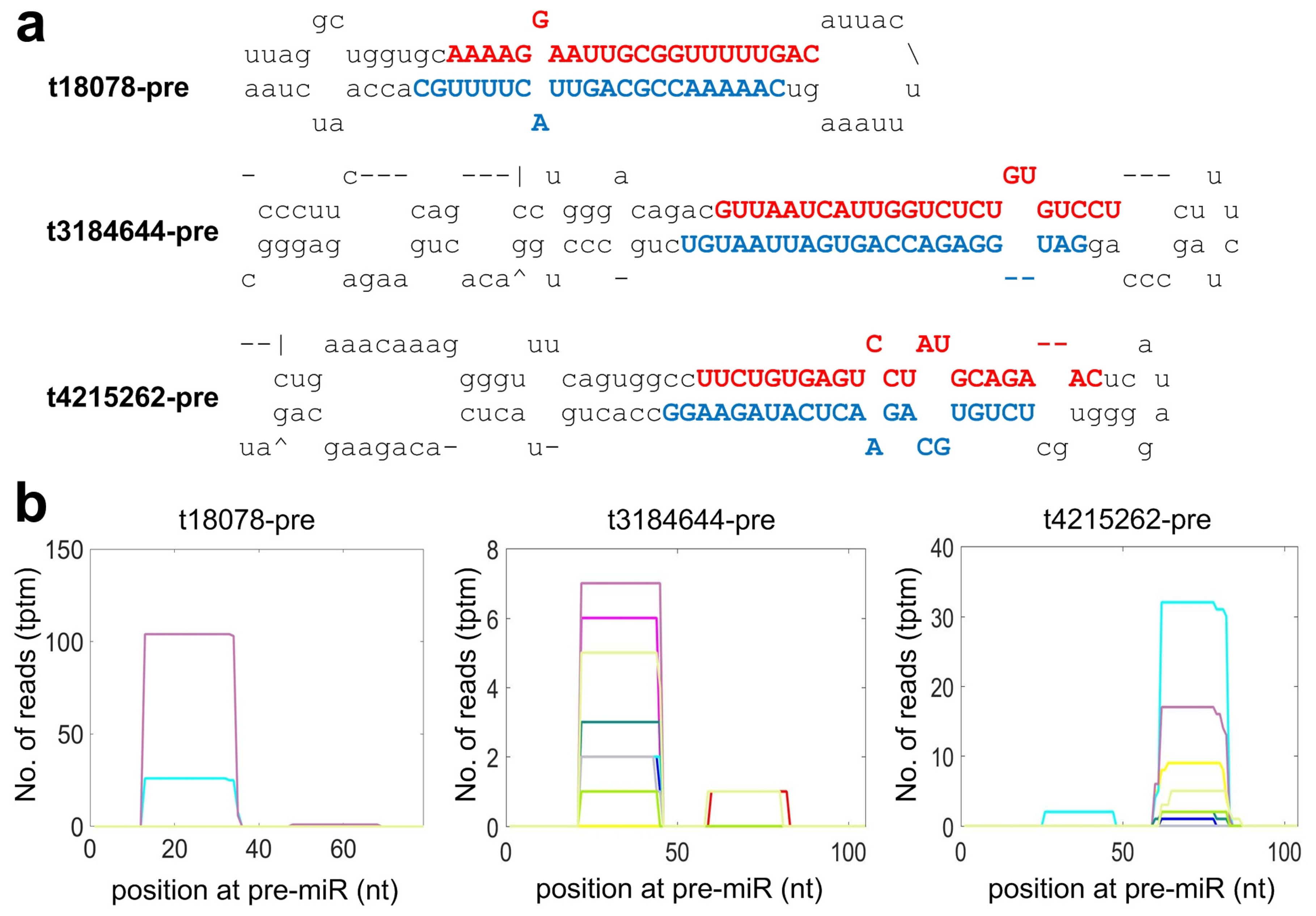
2.4. Identifying Species Specific miRNAs of Macaca mulatta
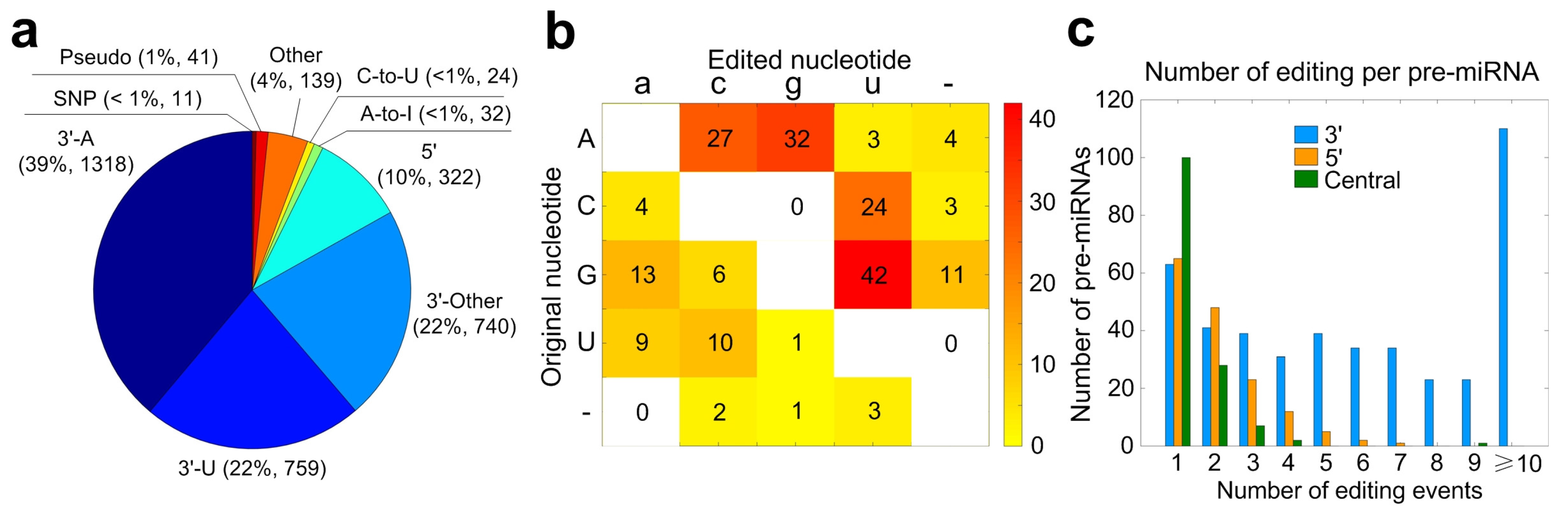
2.5. Summary of Mutation and/or Editing Sites in miRNAs of Macaca mulatta
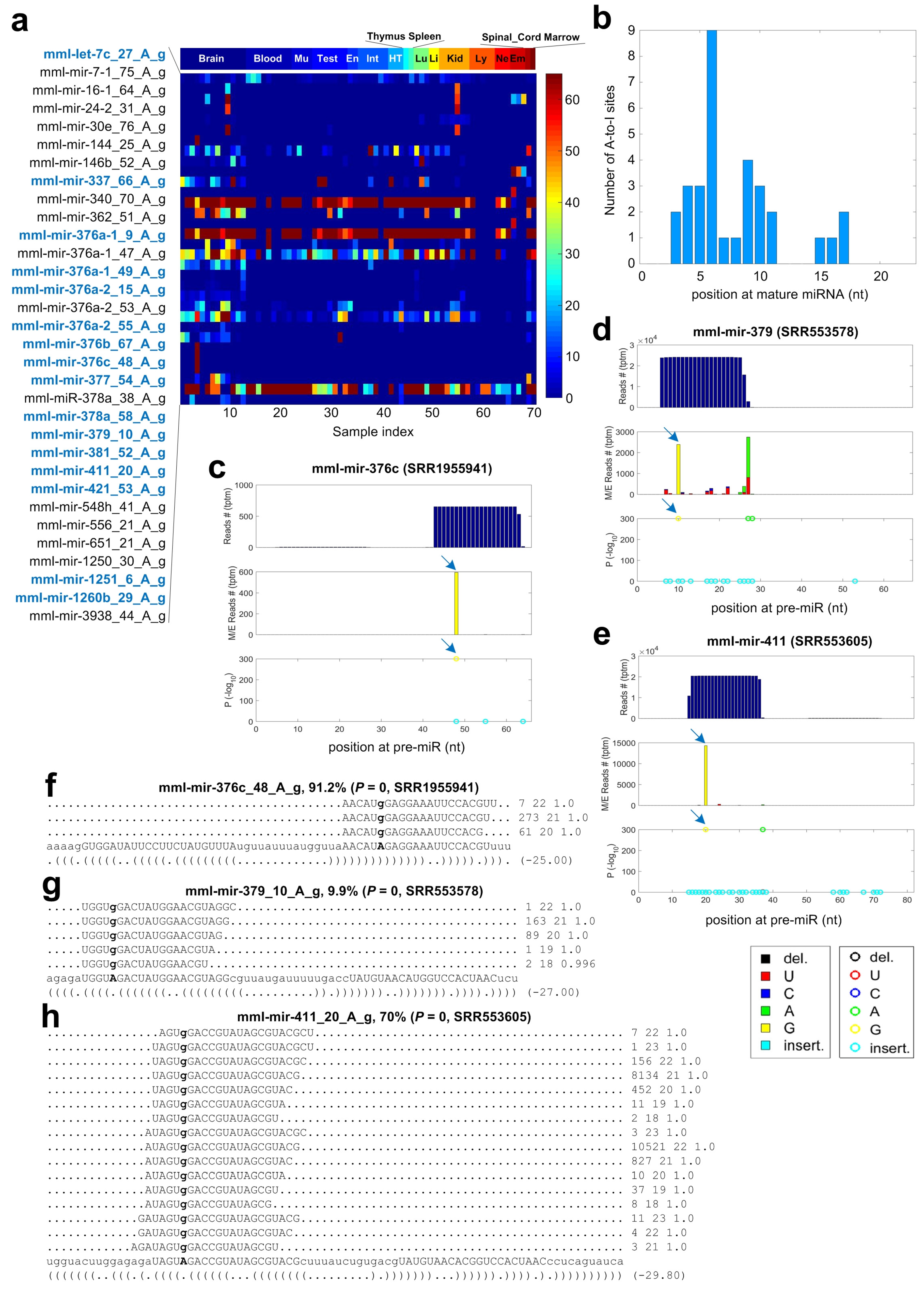
2.6. A-to-I Editing Sites in miRNAs of Macaca mulatta
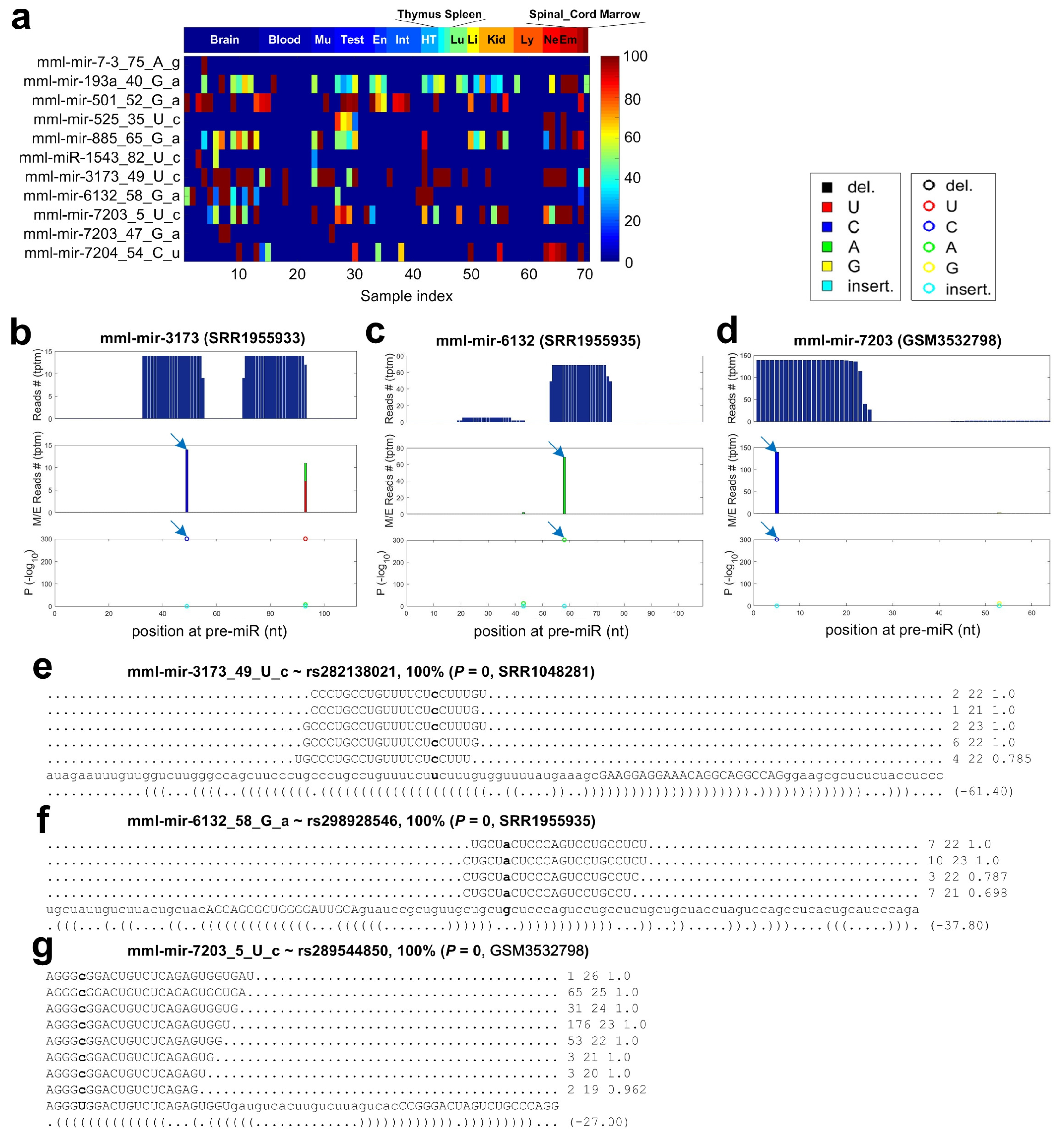
2.7. C-to-U Editing Sites in miRNAs of Macaca mulatta
2.8. Non-Canonical Editing Sites in miRNAs of Macaca mulatta
2.9. Identified SNPs in miRNAs of Macaca mulatta
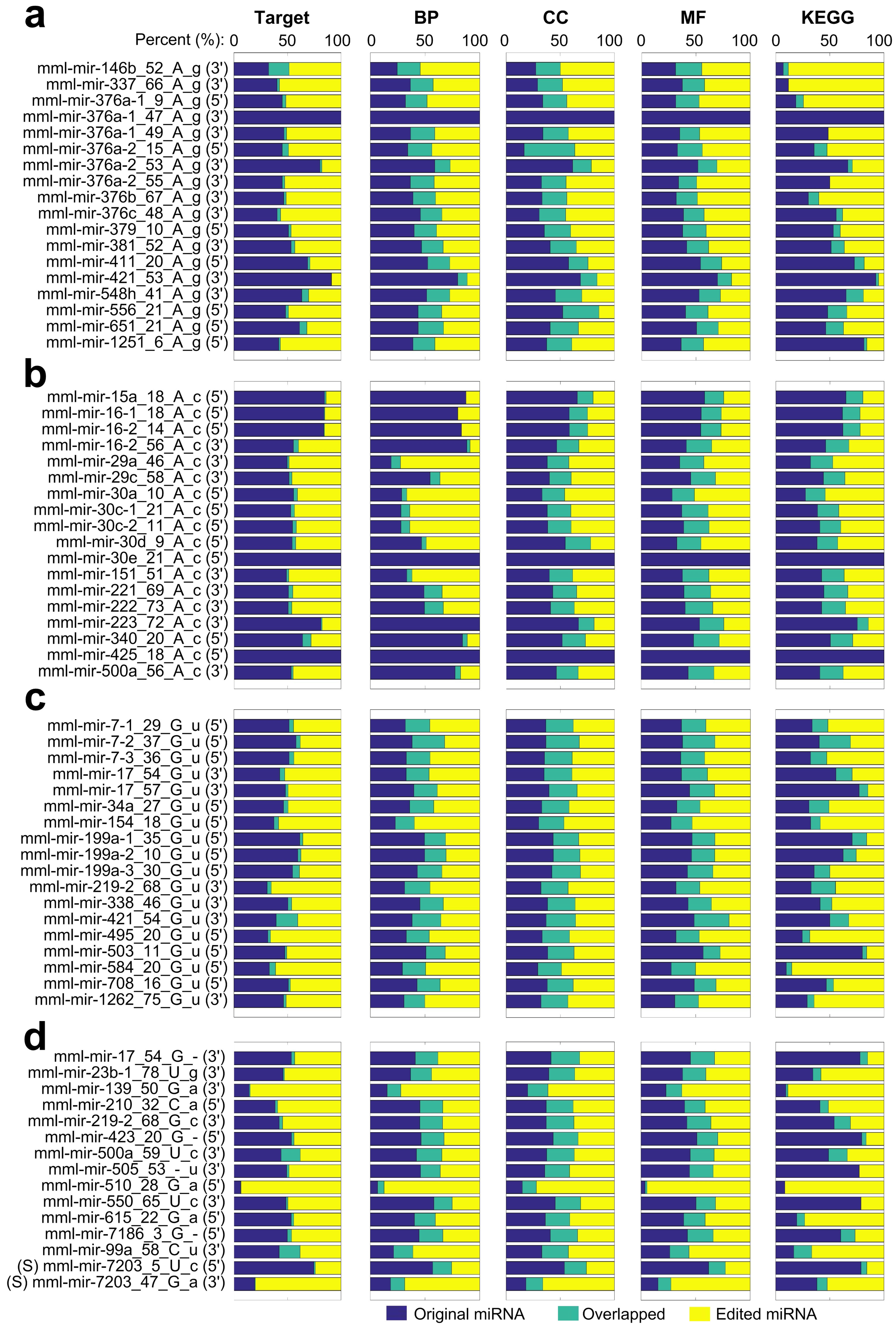
2.10. The Potential Functions of M/E Sites Identified in miRNAs of Macaca mulatta
3. Materials and Methods
3.1. Materials and Small RNA Sequencing Profiles
3.2. Identification of Conserved miRNAs
3.3. Analysis of the Expression Patterns of Conserved miRNAs
3.4. Identification of Species Specific miRNAs
3.5. Identifying Editing and Mutation Sites in miRNAs
3.6. Predicting Putative Targets for the Original and Changed miRNAs
3.7. Go and Pathway Analysis for the Original and Changed miRNAs
4. Conclusions
5. Declarations
5.1. Ethics Approval
5.2. Availability of Data and Materials
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Comai, L.; Zhang, B. MicroRNAs: Key gene regulators with versatile functions. Plant Mol. Biol. 2012, 80, 1. [Google Scholar] [CrossRef] [PubMed]
- Lai, V.K.; Ashraf, M.; Jiang, S.; Haider, K. MicroRNA-143 is a critical regulator of cell cycle activity in stem cells with co-overexpression of Akt and angiopoietin-1 via transcriptional regulation of Erk5/cyclin D1 signaling. Cell Cycle 2012, 11, 767–777. [Google Scholar] [PubMed] [Green Version]
- Ng, R.; Song, G.; Roll, G.R.; Frandsen, N.M.; Willenbring, H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Investig. 2012, 122, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Hans-Ingo, T.; Janine, D.; Eugenie, H.; Iwaniuk, K.M.; Markus, H.; Neil, R.; Thomas, T.; Peter, W. MicroRNAs miR-26a, miR-26b, and miR-29b accelerate osteogenic differentiation of unrestricted somatic stem cells from human cord blood. BMC Genom. 2013, 14, 111. [Google Scholar]
- Tong, M.H.; Mitchell, D.A.; Mcgowan, S.D.; Evanoff, R.; Griswold, M.D. Two miRNA Clusters, Mir-17 (Mirc1) Mir-106b (Mirc3), Are Involv. Regul. Spermatogonial Differ. Mice. Biol. Reprod. 2012, 86, 72. [Google Scholar] [CrossRef]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Slack, F. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Lu, M.; Zhang, Q.; Deng, M.; Miao, J.; Guo, Y.; Gao, W.; Cui, Q. An analysis of human microRNA and disease associations. PLoS ONE 2008, 3, e3420. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.X.; Yan, G.Y. RWRMDA: Predicting novel human microRNA-disease associations. Mol. Biosyst. 2012, 8, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Rooij, E.V.; Olson, E.N. MicroRNA therapeutics for cardiovascular disease: Opportunities and obstacles. Nat. Rev. Drug Discov. 2012, 11, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Bass, B.; Nishikura, K.; Keller, W.; Seeburg, P.; Emeson, R.; O’connell, M.; Samuel, C.; Herbert, A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 1997, 3, 947. [Google Scholar] [PubMed]
- Luciano, D.J.; Mirsky, H.; Vendetti, N.J.; Maas, S. RNA editing of a miRNA precursor. RNA 2004, 10, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.; Grocock, R.; Van Dongen, S.; Enright, A.; Dicks, E.; Futreal, P.; Wooster, R.; Stratton, M. RNA editing of human microRNAs. Genome Biol. 2006, 7, R27. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef]
- Kawahara, Y.; Megraw, M.; Kreider, E.; Iizasa, H.; Valente, L.; Hatzigeorgiou, A.; Nishikura, K. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008, 36, 5270–5280. [Google Scholar] [CrossRef]
- De Hoon, M.J.L.; Taft, R.J.; Hashimoto, T.; Kanamori-Katayama, M.; Kawaji, H.; Kawano, M.; Kishima, M.; Lassmann, T.; Faulkner, G.J.; Mattick, J.S.; et al. Cross-mapping and the identification of editing sites in mature microRNAs in high-throughput sequencing libraries. Genome Res. 2010, 20, 257–264. [Google Scholar] [CrossRef] [Green Version]
- Burroughs, A.M.; Ando, Y.; de Hoon, M.J.L.; Tomaru, Y.; Nishibu, T.; Ukekawa, R.; Funakoshi, T.; Kurokawa, T.; Suzuki, H.; Hayashizaki, Y.; et al. A comprehensive survey of 3’ animal miRNA modification events and a possible role for 3’ adenylation in modulating miRNA targeting effectiveness. Genome Res. 2010, 20, 1398–1410. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Q.; Lu, J.; Li, H.; Ge, Q.; Gu, W.; Bai, Y.; Lu, Z. A comprehensive survey of miRNA repertoire and 3’ addition events in the placentas of patients with pre-eclampsia from high-throughput sequencing. PLoS ONE 2011, 6, e21072. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.; Knouf, E.; Parkin, R.; Fritz, B.; Lin, D.; Dennis, L.; Krouse, M.; Webster, P.; Tewari, M. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011, 21, 1450–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuguchi, Y.; Mishima, T.; Yokomuro, S.; Arima, Y.; Kawahigashi, Y.; Shigehara, K.; Kanda, T.; Yoshida, H.; Uchida, E.; Tajiri, T.; et al. Sequencing and Bioinformatics-Based Analyses of the microRNA Transcriptome in Hepatitis B–Related Hepatocellular Carcinoma. PLoS ONE 2011, 6, e15304. [Google Scholar] [CrossRef] [PubMed]
- Alon, S.; Mor, E.; Vigneault, F.; Church, G.M.; Locatelli, F.; Galeano, F.; Gallo, A.; Shomron, N.; Eisenberg, E. Systematic identification of edited microRNAs in the human brain. Genome Res. 2012, 22, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekdahl, Y.; Farahani, H.; Behm, M.; Lagergren, J.; Öhman, M. A-to-I editing of microRNAs in the mammalian brain increases during development. Genome Res. 2012, 22, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.J.J.; Park, J.E.E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-Uridylation of Pre-MicroRNA as a Key Step in the Biogenesis of Group II let-7 MicroRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of Silencing Targets by Adenosine-to-Inosine Editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2005, 13, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y.; Zinshteyn, B.; Chendrimada, T.P.; Shiekhattar, R.; Nishikura, K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007, 8, 763–769. [Google Scholar] [CrossRef]
- Vesely, C.; Tauber, S.; Sedlazeck, F.J.; von Haeseler, A.; Jantsch, M.F. Adenosine deaminases that act on RNA induce reproducible changes in abundance and sequence of embryonic miRNAs. Genome Res. 2012, 22, 1468–1476. [Google Scholar] [CrossRef] [Green Version]
- Park, E.; Williams, B.; Wold, B.J.; Mortazavi, A. RNA editing in the human ENCODE RNA-seq data. Genome Res. 2012, 22, 1626–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, V.; Paul, D.; Das, S.; Bajpai, P.; Singh, S.; Mukhopadhyay, A.; Agrawal, A.; Ghosh, B. Altered expression and editing of miRNA-100 regulates iTreg differentiation. Nucleic Acids Res. 2015, 43, 8057–8065. [Google Scholar] [CrossRef] [PubMed]
- Ichinose, M.; Sugita, M. RNA Editing and Its Molecular Mechanism in Plant Organelles. Genes 2016, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Gagnidze, K.; Rayon-Estrada, V.; Harroch, S.; Bulloch, K.; Nina Papavasiliou, F. A New Chapter in Genetic Medicine: RNA Editing and its Role in Disease Pathogenesis. Trends Mol. Med. 2018, 24. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Li, T.; Ren, R.; Shi, D.; Wang, S. Revealing editing and SNPs of microRNAs in colon tissues by analyzing high-throughput sequencing profiles of small RNAs. BMC Genom. 2014, 15, S11. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ji, B.; Song, R.; Wang, S.; Li, T.; Zhang, X.; Chen, K.; Li, T.; Li, J. Accurate detection for a wide range of mutation and editing sites of microRNAs from small RNA high-throughput sequencing profiles. Nucleic Acids Res. 2016, 44, e123. [Google Scholar] [CrossRef]
- Morin, R.D.; O’Connor, M.D.; Griffith, M.; Kuchenbauer, F.; Delaney, A.; Prabhu, A.L.L.; Zhao, Y.; McDonald, H.; Zeng, T.; Hirst, M.; et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res. 2008, 18, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.K.; Heo, I.; Kim, V.N. Modifications of small RNAs and their associated proteins. Cell 2010, 143, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Pak, C.; Jin, P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum. Mol. Genet. 2007, 16, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.; Robles, A.; Harris, C. Genetic variation in microRNA networks: The implications for cancer research. Nat. Rev. Cancer 2010, 10, 389–402. [Google Scholar] [CrossRef]
- Calin, G.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.; Iorio, M.; Visone, R.; Sever, N.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Shoshan, E.; Mobley, A.K.; Braeuer, R.R.; Kamiya, T.; Huang, L.; Vasquez, M.E.; Salameh, A.; Lee, H.J.; Kim, S.J.; Ivan, C.; et al. Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis. Nat. Cell Biol. 2015, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Rogers, J.; Katze, M.G.; Bumgarner, R.; Weinstock, G.M.; Mardis, E.R.; Remington, K.A.; Strausberg, R.L.; Venter, J.C.; Wilson, R.K.; et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science 2007, 316, 222–234. [Google Scholar] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y. Computational Non-Coding RNA Biology, 1st ed.; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Chen, J.; Mandel, E.; Thomson, J.; Wu, Q.; Callis, T.; Hammond, S.; Conlon, F.; Wang, D. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhou, Y.; Jiang, L.; Li, D.; Yang, J.; Zhang, C.Y.; Zen, K. Urinary MicroRNA-10a and MicroRNA-30d Serve as Novel, Sensitive and Specific Biomarkers for Kidney Injury. PLoS ONE 2012, 7, e51140. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Meyers, B.C. Revisiting criteria for plant miRNA annotation in the era of big data. Plant Cell 2018, 30, 272–284. [Google Scholar] [CrossRef]
- Nigita, G.; Acunzo, M.; Romano, G.; Veneziano, D.; Laganà, A.; Vitiello, M.; Wernicke, D.; Ferro, A.; Croce, C.M. MicroRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 2016, 44, gkw532. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, T.; Xu, Z.; Wai, C.M.; Chen, K.; Zhang, X.; Wang, S.; Ji, B.; Ming, R.; Sunkar, R. Identification of microRNAs, phasiRNAs and Their Targets in Pineapple. Trop. Plant Biol. 2016, 9, 176–186. [Google Scholar] [CrossRef]
- Liu, L.; Ren, S.; Guo, J.; Wang, Q.; Zhang, X.; Liao, P.; Li, S.; Sunkar, R.; Zheng, Y. Genome-wide identification and comprehensive analysis of microRNAs and phased small interfering RNAs in watermelon. BMC Genom. 2018, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, C.; Li, Y.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Burge, S.W.; Daub, J.; Eberhardt, R.; Tate, J.; Barquist, L.; Nawrocki, E.P.; Eddy, S.R.; Gardner, P.P.; Bateman, A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013, 41, D226–D232. [Google Scholar] [CrossRef]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbø, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3. 0: Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 1. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Kiran, A.; Baranov, P.V. DARNED: A DAtabase of RNa EDiting in humans. Bioinformatics 2010, 26, 1772–1776. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Li, J.B. RADAR: A rigorously annotated database of A-to-I RNA editing. Nucleic Acids Res. 2014, 42, D109. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Wu, Y.; Zhang, X.; Liao, Y.; Sibanda, V.L.; Liu, W.; Guo, A.Y. Comprehensive analysis of human small RNA sequencing data provides insights into expression profiles and miRNA editing. RNA Biol. 2014, 11, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Zhang, W. Animal microRNA Target Prediction Using Diverse Sequence-Specific Determinants. J. Bioinform. Comput. Biol. 2010, 8, 763–788. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhao, Z.; Zhang, X.; Lu, C.; Ren, S.; Li, S.; Guo, J.; Liao, P.; Jiang, B.; Zheng, Y. Identifying microRNAs and Their Editing Sites in Macaca mulatta. Cells 2019, 8, 682. https://doi.org/10.3390/cells8070682
Wang Q, Zhao Z, Zhang X, Lu C, Ren S, Li S, Guo J, Liao P, Jiang B, Zheng Y. Identifying microRNAs and Their Editing Sites in Macaca mulatta. Cells. 2019; 8(7):682. https://doi.org/10.3390/cells8070682
Chicago/Turabian StyleWang, Qingyi, Zhigang Zhao, Xiaotuo Zhang, Chenyu Lu, Shuchao Ren, Shipeng Li, Junqiang Guo, Peiran Liao, Bingbing Jiang, and Yun Zheng. 2019. "Identifying microRNAs and Their Editing Sites in Macaca mulatta" Cells 8, no. 7: 682. https://doi.org/10.3390/cells8070682
APA StyleWang, Q., Zhao, Z., Zhang, X., Lu, C., Ren, S., Li, S., Guo, J., Liao, P., Jiang, B., & Zheng, Y. (2019). Identifying microRNAs and Their Editing Sites in Macaca mulatta. Cells, 8(7), 682. https://doi.org/10.3390/cells8070682