Interplay between Zika Virus and Peroxisomes during Infection


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus Infection
2.2. Plasmids and Transfection
2.3. Production of Lentiviral Particles and Transduction of Cells
2.4. Antibodies
2.5. Immunoblotting
2.6. Quantitative Real-Time PCR (qRT-PCR)
2.7. Co-Immunoprecipitation and Immunoblotting
2.8. Cell Viability Assay
2.9. Confocal Microscopy
2.10. Quantification of Peroxisomes
2.11. Statistical Analyses
3. Results
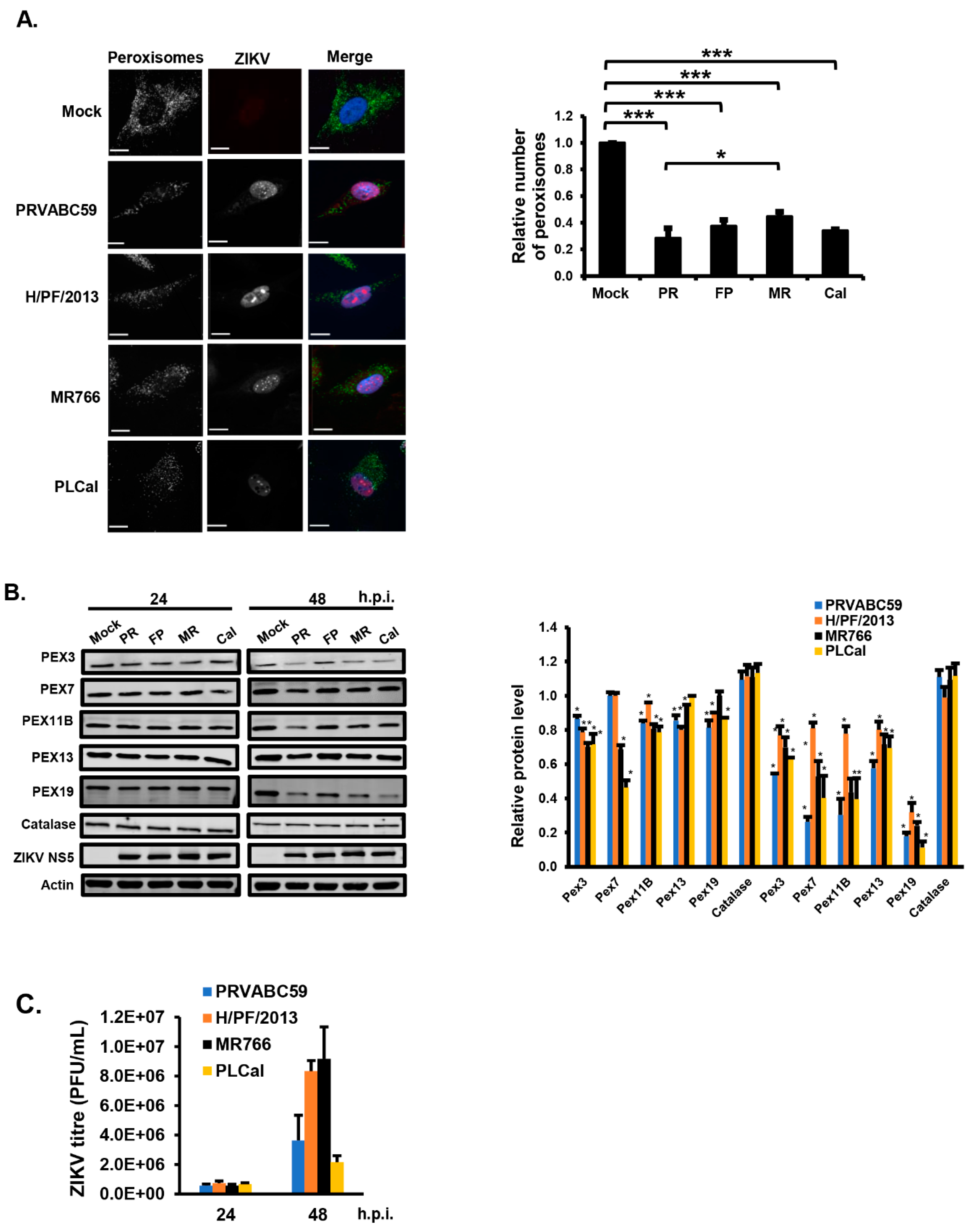
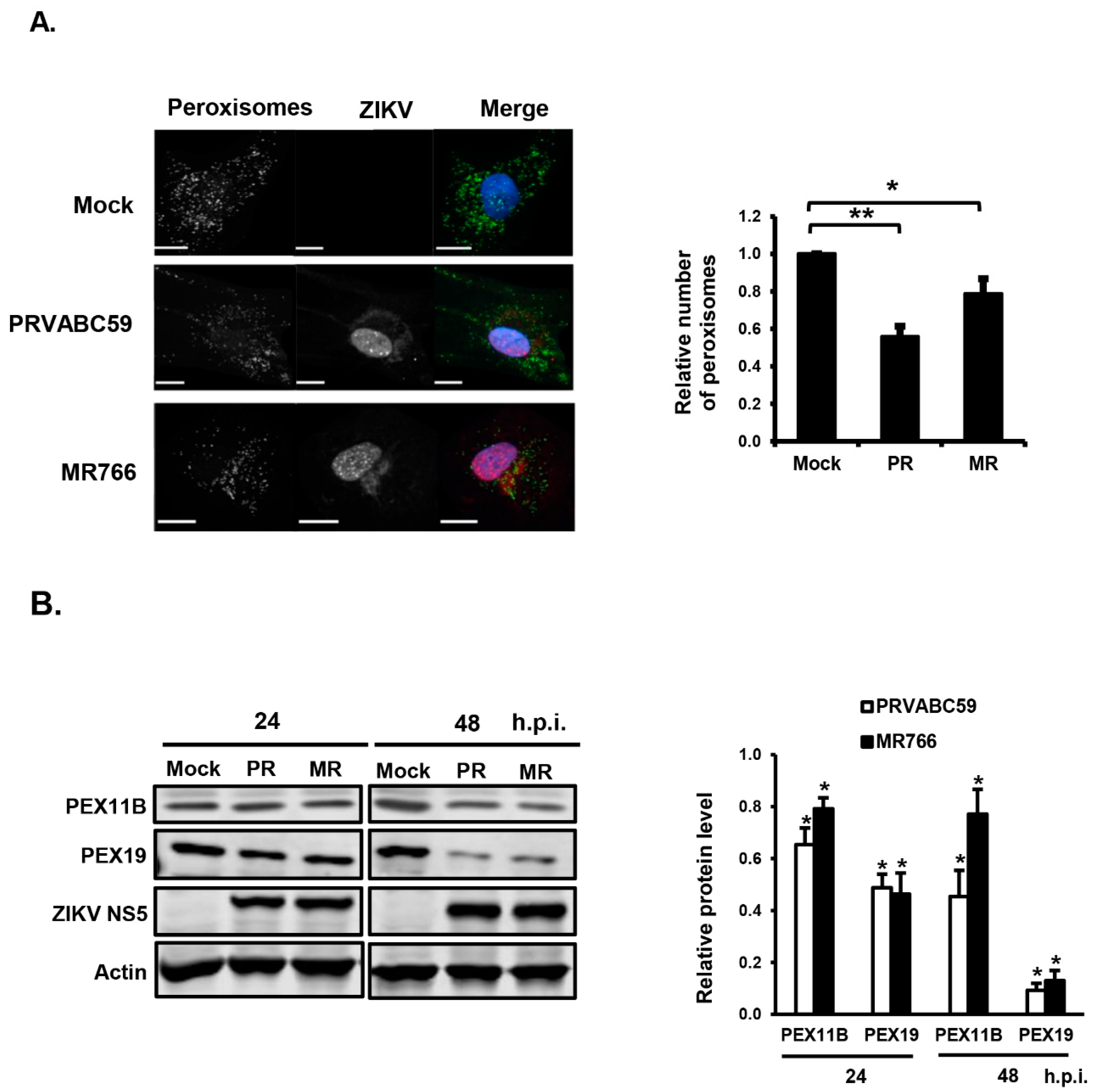
3.1. ZIKV Infection Decreases the Pool of Peroxisomes in Primary HFAs and U251 Cells
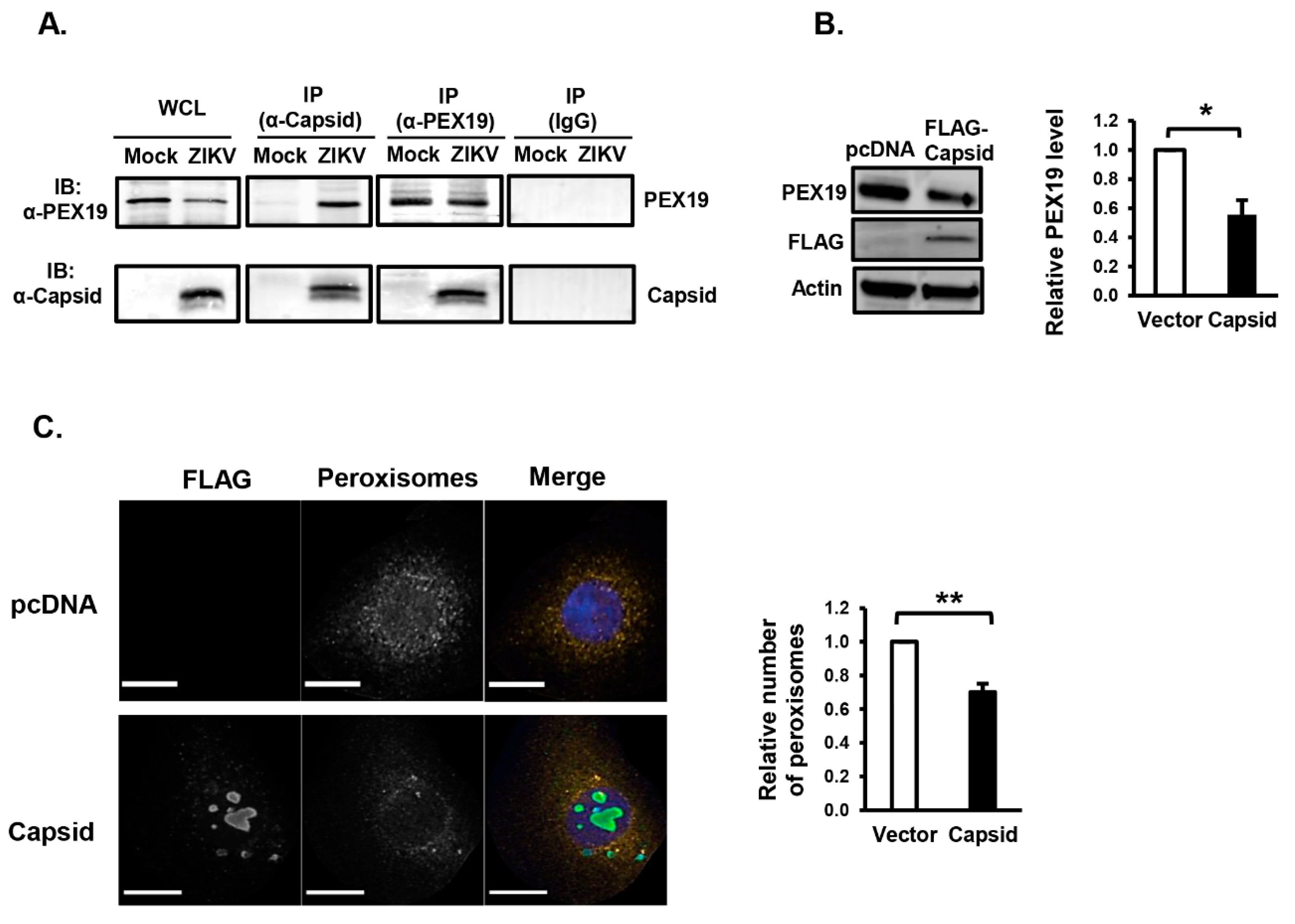
3.2. ZIKV Capsid Protein Binds PEX19 and Induces Loss of Peroxisomes
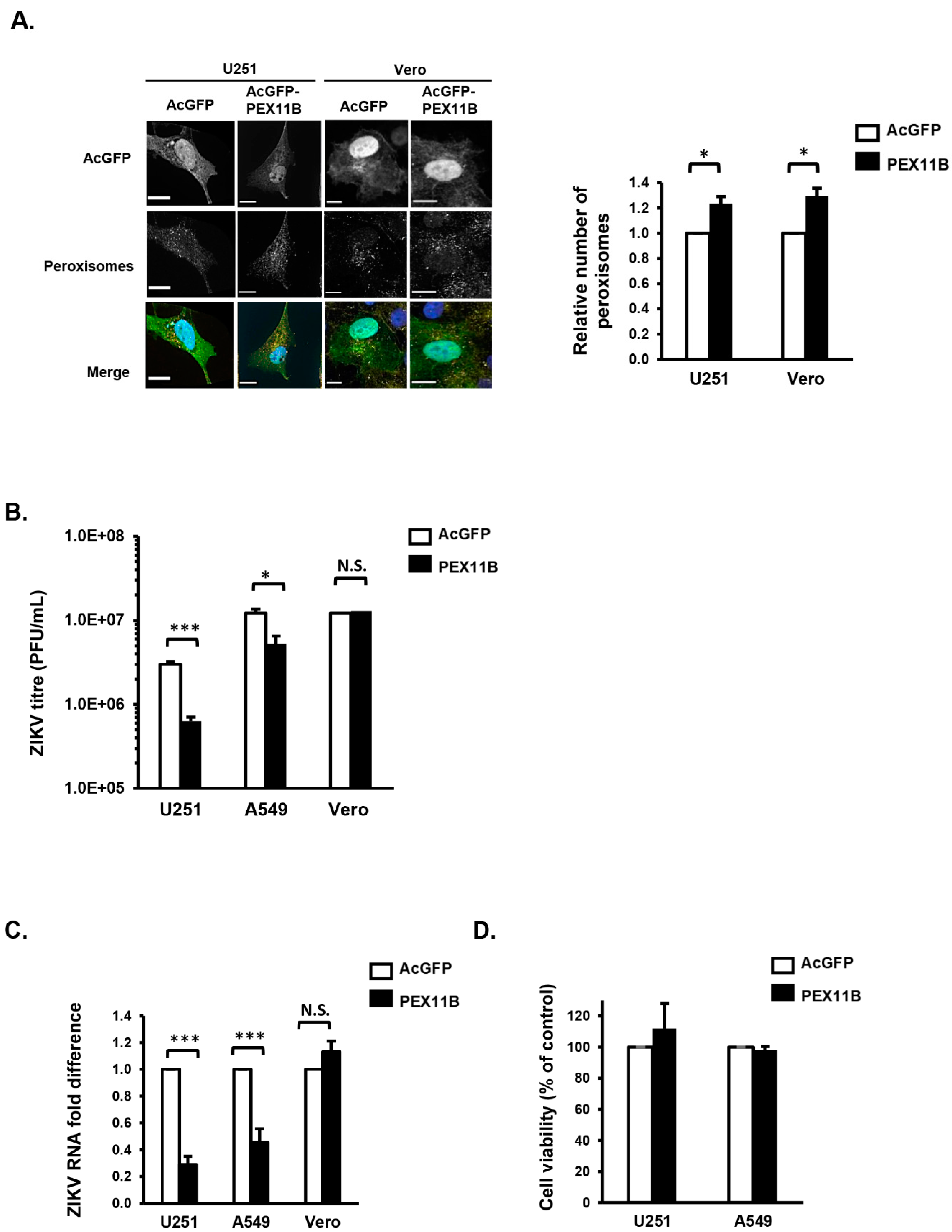
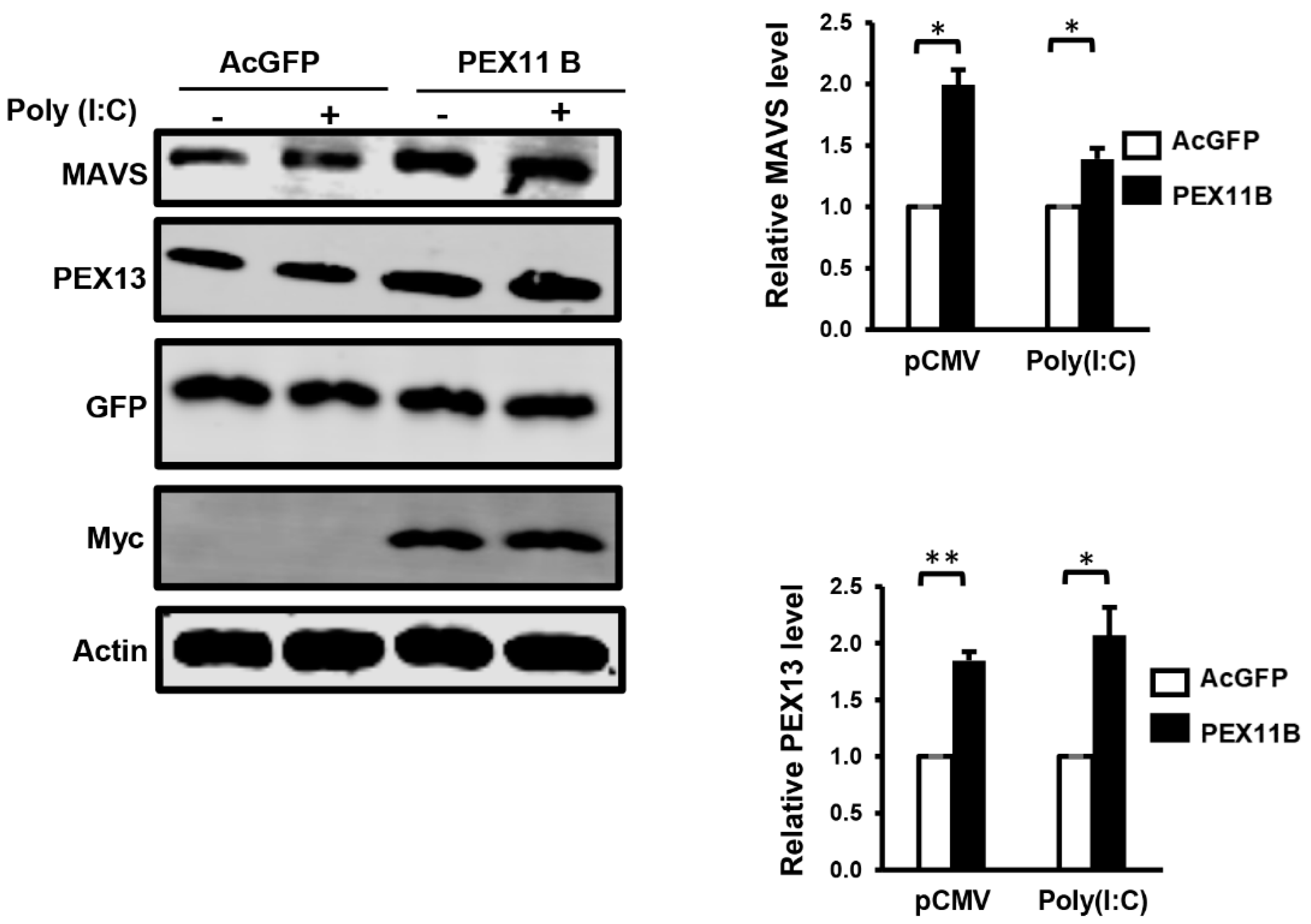
3.3. Over-Expression of PEX11B Inhibits ZIKV Replication
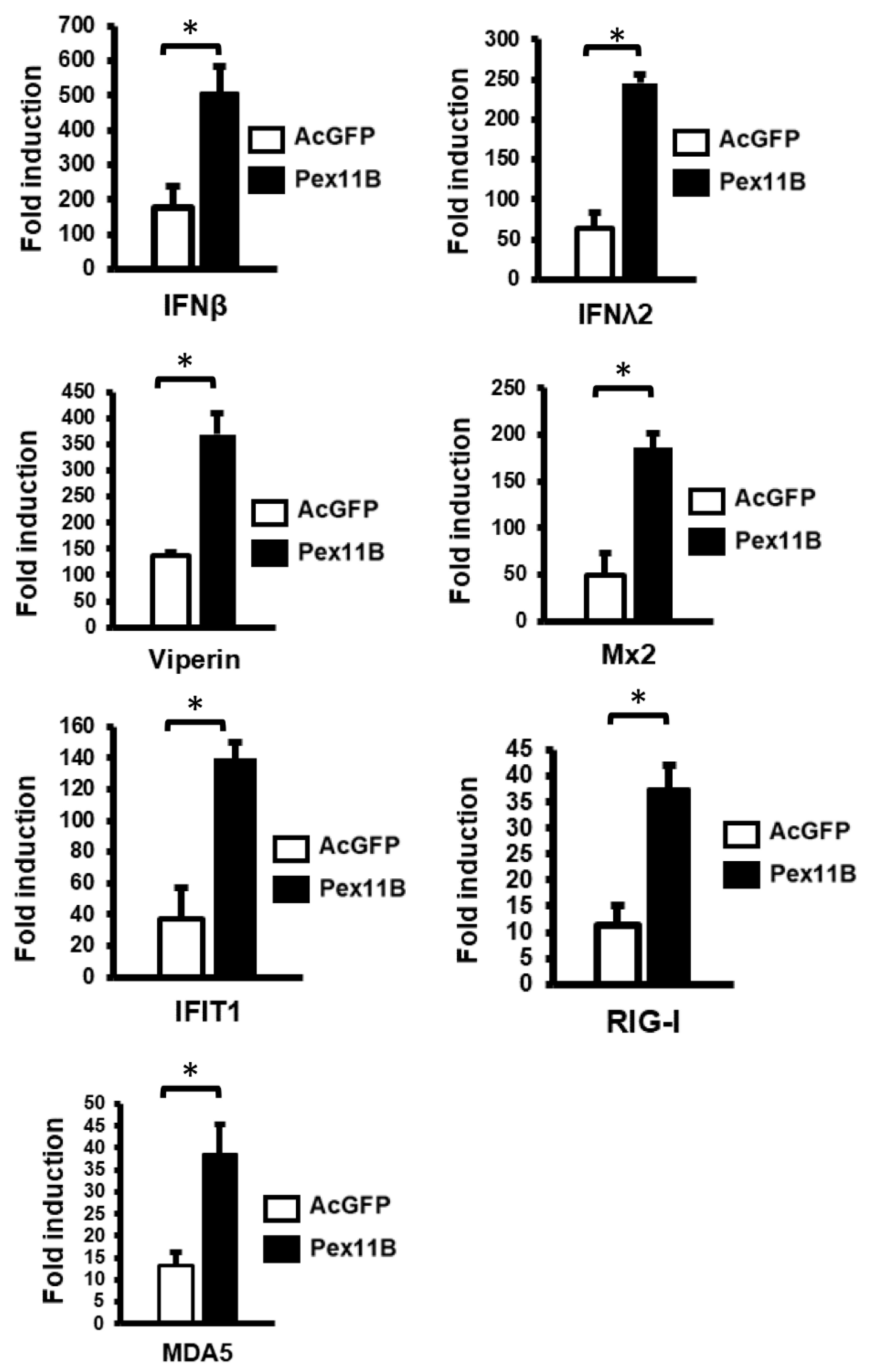
3.4. Over-Expression of PEX11B Enhances the Innate Immune Response
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kuno, G.; Chang, G.J. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch. Virol. 2007, 152, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.A.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Rubin, E.J.; Greene, M.F.; Baden, L.R. Zika Virus and Microcephaly. N. Engl. J. Med. 2016, 374, 984–985. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.C. Microcephaly and Zika virus infection. Lancet 2016, 387, 2070–2072. [Google Scholar] [CrossRef] [Green Version]
- Yockey, L.J.; Varela, L.; Rakib, T.; Khoury-Hanold, W.; Fink, S.L.; Stutz, B.; Szigeti-Buck, K.; Van den Pol, A.; Lindenbach, B.D.; Horvath, T.L.; et al. Vaginal Exposure to Zika Virus during Pregnancy Leads to Fetal Brain Infection. Cell 2016, 166, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Aliota, M.T.; Caine, E.A.; Walker, E.C.; Larkin, K.E.; Camacho, E.; Osorio, J.E. Characterization of Lethal Zika Virus Infection in AG129 Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004682. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef]
- Kell, A.M.; Gale, M. RIG-I in RNA virus recognition. Virology 2015, 479, 110–121. [Google Scholar] [CrossRef]
- Pichlmair, A.; e Sousa, C.R. Innate recognition of viruses. Immunity 2007, 27, 370–383. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marié, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Hughes, R.; Towers, G.; Noursadeghi, M. Innate immune interferon responses to human immunodeficiency virus-1 infection. Rev. Med. Virol. 2012, 22, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Voelkl, A.; Fahimi, H.D.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histochem. Cell Biol. 2018, 150, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Deori, N.M.; Kale, A.; Maurya, P.K.; Nagotu, S. Peroxisomes: Role in cellular ageing and age related disorders. Biogerontology 2018, 19, 303–324. [Google Scholar] [CrossRef] [PubMed]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Bender, S.; Reuter, A.; Eberle, F.; Einhorn, E.; Binder, M.; Bartenschlager, R. Activation of Type I and III Interferon Response by Mitochondrial and Peroxisomal MAVS and Inhibition by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005264. [Google Scholar] [CrossRef]
- Dixit, E.; Boulant, S.; Zhang, Y.; Lee, A.S.; Odendall, C.; Shum, B.; Hacohen, N.; Chen, Z.J.; Whelan, S.P.; Fransen, M.; et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell 2010, 141, 668–681. [Google Scholar] [CrossRef]
- You, J.; Hou, S.; Malik-Soni, N.; Xu, Z.; Kumar, A.; Rachubinski, R.A.; Frappier, L.; Hobman, T.C. Flavivirus Infection Impairs Peroxisome Biogenesis and Early Antiviral Signaling. J. Virol. 2015, 89, 12349–12361. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.R.; Magalhães, A.C.; Camões, F.; Gouveia, A.; Vieira, M.; Kagan, J.C.; Ribeiro, D. Hepatitis C virus NS3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J. Cell Mol. Med. 2016, 20, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ke, H.; Blikslager, A.; Fujita, T.; Yoo, D. Type III interferon restriction by porcine epidemic diarrhea virus and the role of viral protein nsp1 in IRF1 signaling. J. Virol. 2018, 92, e01677-17. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Asahchop, E.L.; Branton, W.G.; Gelman, B.B.; Power, C.; Hobman, T.C. MicroRNAs upregulated during HIV infection target peroxisome biogenesis factors: Implications for virus biology, disease mechanisms and neuropathology. PLoS Pathog. 2017, 13, e1006360. [Google Scholar] [CrossRef] [PubMed]
- Coyaud, E.; Ranadheera, C.; Cheng, D.; Gonçalves, J.; Dyakov, B.J.A.; Laurent, E.M.N.; St-Germain, J.; Pelletier, L.; Gingras, A.C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell Proteomics 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018, 10, 646. [Google Scholar] [CrossRef] [PubMed]
- Kassmann, C.M.; Lappe-Siefke, C.; Baes, M.; Brügger, B.; Mildner, A.; Werner, H.B.; Natt, O.; Michaelis, T.; Prinz, M.; Frahm, J.; et al. Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes. Nat. Genet. 2007, 39, 969–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bottelbergs, A.; Verheijden, S.; Hulshagen, L.; Gutmann, D.H.; Goebbels, S.; Nave, K.A.; Kassmann, C.; Baes, M. Axonal integrity in the absence of functional peroxisomes from projection neurons and astrocytes. Glia 2010, 58, 1532–1543. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Schwarz, M.C.; Sourisseau, M.; Espino, M.M.; Gray, E.S.; Chambers, M.T.; Tortorella, D.; Evans, M.J. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. MSphere 2016, 1, e00246-16. [Google Scholar] [CrossRef]
- Urbanowski, M.D.; Hobman, T.C. The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Götte, M.; Power, C. Zika virus hijacks stress granule proteins and modulates the host stress response. J. Virol. 2017, 91, e00474-17. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Shepard, D.S.; Halasa, Y.A.; Tyagi, B.K.; Adhish, S.V.; Nandan, D.; Karthiga, K.S.; Chellaswamy, V.; Gaba, M.; Arora, N.K.; Group, I.S. Economic and disease burden of dengue illness in India. Am. J. Trop. Med. Hyg. 2014, 91, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Gould, S.J.; Keller, G.A.; Hosken, N.; Wilkinson, J.; Subramani, S. A conserved tripeptide sorts proteins to peroxisomes. J. Cell Biol. 1989, 108, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Schrader, M.; Reuber, B.E.; Morrell, J.C.; Jimenez-Sanchez, G.; Obie, C.; Stroh, T.A.; Valle, D.; Schroer, T.A.; Gould, S.J. Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli. J. Biol. Chem. 1998, 273, 29607–29614. [Google Scholar] [CrossRef] [PubMed]
- Opaliński, Ł.; Kiel, J.A.; Williams, C.; Veenhuis, M.; van der Klei, I.J. Membrane curvature during peroxisome fission requires Pex11. EMBO J. 2011, 30, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Desmyter, J.; Melnick, J.L.; Rawls, W.E. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 1968, 2, 955–961. [Google Scholar]
- Helbig, K.J.; Lau, D.T.; Semendric, L.; Harley, H.A.; Beard, M.R. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 2005, 42, 702–710. [Google Scholar] [CrossRef]
- Helbig, K.J.; Eyre, N.S.; Yip, E.; Narayana, S.; Li, K.; Fiches, G.; McCartney, E.M.; Jangra, R.K.; Lemon, S.M.; Beard, M.R. The antiviral protein viperin inhibits hepatitis C virus replication via interaction with nonstructural protein 5A. Hepatology 2011, 54, 1506–1517. [Google Scholar] [CrossRef]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Negl. Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.S.; Vonderstein, K.; Pichlmair, A.; Stehling, O.; Bennett, K.L.; Dobler, G.; Guo, J.T.; Superti-Furga, G.; Lill, R.; Överby, A.K.; et al. Viperin is an iron-sulfur protein that inhibits genome synthesis of tick-borne encephalitis virus via radical SAM domain activity. Cell Microbiol. 2014, 16, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoek, K.H.; Eyre, N.S.; Shue, B.; Khantisitthiporn, O.; Glab-Ampi, K.; Carr, J.M.; Gartner, M.J.; Jolly, L.A.; Thomas, P.Q.; Adikusuma, F.; et al. Viperin is an important host restriction factor in control of Zika virus infection. Sci. Rep. 2017, 7, 4475. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. MBio 2015, 6, e00553-15. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef]
- Wong, C.P.; Xu, Z.; Power, C.; Hobman, T.C. Targeted Elimination of Peroxisomes During Viral Infection: Lessons from HIV and Other Viruses. DNA Cell Biol. 2018, 37, 417–421. [Google Scholar] [CrossRef]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Li, Y.; Zhang, H.; Zhao, R.; Jing, R.; Xu, Y.; He, M.; Peer, J.; Kim, Y.C.; Luo, J.; et al. Zika virus propagation and release in human fetal astrocytes can be suppressed by neutral sphingomyelinase-2 inhibitor GW4869. Cell Discov. 2018, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Limonta, D.; Jovel, J.; Kumar, A.; Lu, J.; Hou, S.; Airo, A.M.; Lopez-Orozco, J.; Wong, C.P.; Saito, L.; Branton, W.; et al. Fibroblast growth factor 2 enhances Zika virus infection in human fetal brain. J. Infect. Dis. 2019. [Google Scholar] [CrossRef] [PubMed]
- Beltran, P.M.J.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell Host Microbe 2018, 24, 526–541. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, C.P.; Xu, Z.; Hou, S.; Limonta, D.; Kumar, A.; Power, C.; Hobman, T.C. Interplay between Zika Virus and Peroxisomes during Infection. Cells 2019, 8, 725. https://doi.org/10.3390/cells8070725
Wong CP, Xu Z, Hou S, Limonta D, Kumar A, Power C, Hobman TC. Interplay between Zika Virus and Peroxisomes during Infection. Cells. 2019; 8(7):725. https://doi.org/10.3390/cells8070725
Chicago/Turabian StyleWong, Cheung Pang, Zaikun Xu, Shangmei Hou, Daniel Limonta, Anil Kumar, Christopher Power, and Tom C. Hobman. 2019. "Interplay between Zika Virus and Peroxisomes during Infection" Cells 8, no. 7: 725. https://doi.org/10.3390/cells8070725
APA StyleWong, C. P., Xu, Z., Hou, S., Limonta, D., Kumar, A., Power, C., & Hobman, T. C. (2019). Interplay between Zika Virus and Peroxisomes during Infection. Cells, 8(7), 725. https://doi.org/10.3390/cells8070725