Targeting Metabolic Reprogramming in Acute Myeloid Leukemia



Abstract
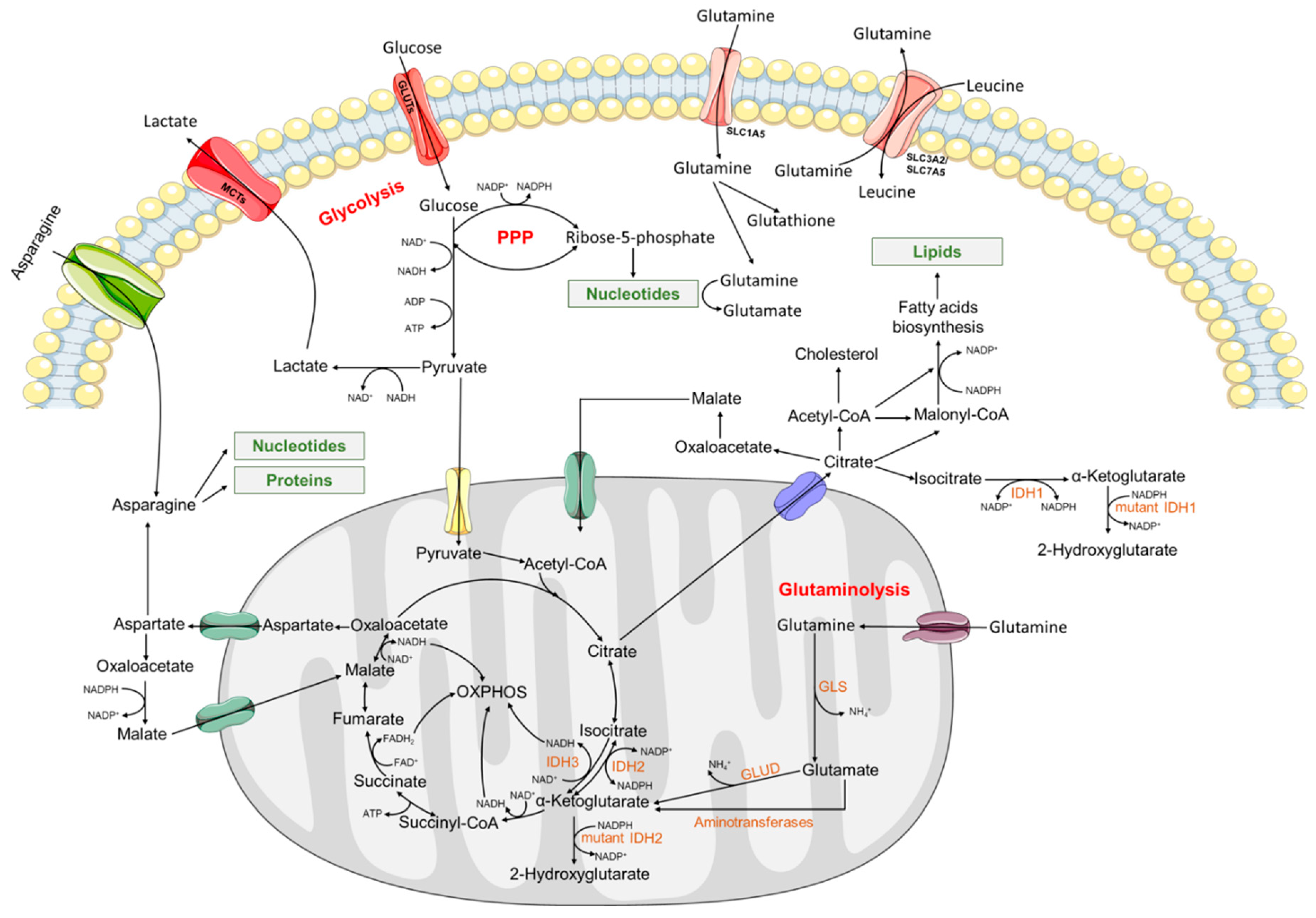
:1. Introduction
2. Metabolic Reprogramming in Acute Myeloid Leukemia
2.1. Glycolysis and Energetic Metabolism
2.2. Amino Acid Metabolism
2.3. Fatty Acid Metabolism
2.4. Metabolic Enzymes and Epigenetic Modifiers
2.5. Signaling Pathways and Autophagy
3. Modulation of AML Metabolism
3.1. Targeting Glycolysis and Energetic Metabolism
3.2. Targeting Amino Acid Metabolism
3.3. Targeting Fatty Acid Metabolism
3.4. Targeting Metabolic Enzymes and Epigenetic Modifiers
3.5. Targeting Signaling Pathways and Autophagy
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Acute Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. Available online: https://www.nccn.org/professionals/physician_gls/default.aspx (accessed on 24 April 2019).
- Estey, E.H. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018, 93, 1267–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klepin, H.D. Myelodysplastic Syndromes and Acute Myeloid Leukemia in the Elderly. Clin. Geriatr. Med. 2016, 32, 155–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Klepin, H.D.; Rao, A.V.; Pardee, T.S. Acute myeloid leukemia and myelodysplastic syndromes in older adults. J. Clin. Oncol. 2014, 32, 2541–2552. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Vachhani, P.; Cortes, J.E. Treatment of Relapsed/Refractory Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Danhier, P.; Banski, P.; Payen, V.L.; Grasso, D.; Ippolito, L.; Sonveaux, P.; Porporato, P.E. Cancer metabolism in space and time: Beyond the Warburg effect. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 556–572. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Herst, P.M.; Howman, R.A.; Neeson, P.J.; Berridge, M.V.; Ritchie, D.S. The level of glycolytic metabolism in acute myeloid leukemia blasts at diagnosis is prognostic for clinical outcome. J. Leukoc. Biol. 2011, 89, 51–55. [Google Scholar] [CrossRef]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Barreau, S.; Stuani, L.; Lambert, M.; Palama, T.L.; Chesnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girao, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef]
- Herst, P.M.; Hesketh, E.L.; Ritchie, D.S.; Berridge, M.V. Glycolytic metabolism confers resistance to combined all-trans retinoic acid and arsenic trioxide-induced apoptosis in HL60rho0 cells. Leuk. Res. 2008, 32, 327–333. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.J.; Xuan, L.; Huang, G.N.; Song, X.L.; Liu, Q.F. HIF-1alpha and GLUT1 gene expression is associated with chemoresistance of acute myeloid leukemia. Asian Pac. J. Cancer Prev. 2014, 15, 1823–1829. [Google Scholar] [CrossRef]
- Song, K.; Li, M.; Xu, X.; Xuan, L.I.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol Lett 2016, 12, 334–342. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Alexeev, E.; Nusbacher, N.; Minhajuddin, M.; Stevens, B.M.; Winters, A.C.; Lin, X.; Ashton, J.M.; et al. Subversion of Systemic Glucose Metabolism as a Mechanism to Support the Growth of Leukemia Cells. Cancer Cell 2018, 34, 659–673. [Google Scholar] [CrossRef]
- Chapuis, N.; Poulain, L.; Birsen, R.; Tamburini, J.; Bouscary, D. Rationale for Targeting Deregulated Metabolic Pathways as a Therapeutic Strategy in Acute Myeloid Leukemia. Front. Oncol. 2019. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci Adv. 2016. [Google Scholar] [CrossRef]
- Tan, H.W.S.; Sim, A.Y.L.; Long, Y.C. Glutamine metabolism regulates autophagy-dependent mTORC1 reactivation during amino acid starvation. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basics of cancer metabolism: Developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017, 12, 833–842. [Google Scholar] [CrossRef]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab. 2018, 27, 428–438. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- Ho, T.C.; LaMere, M.; Stevens, B.M.; Ashton, J.M.; Myers, J.R.; O’Dwyer, K.M.; Liesveld, J.L.; Mendler, J.H.; Guzman, M.; Morrissette, J.D.; et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood 2016, 128, 1671–1678. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab 2013, 18, 153–161. [Google Scholar] [CrossRef]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Stuani, L.; Riols, F.; Millard, P.; Sabatier, M.; Batut, A.; Saland, E.; Viars, F.; Tonini, L.; Zaghdoudi, S.; Linares, L.K.; et al. Stable Isotope Labeling Highlights Enhanced Fatty Acid and Lipid Metabolism in Human Acute Myeloid Leukemia. Int J. Mol. Sci. 2018, 19, 3325. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Shi, J.; Fu, H.; Jia, Z.; He, K.; Fu, L.; Wang, W. High Expression of CPT1A Predicts Adverse Outcomes: A Potential Therapeutic Target for Acute Myeloid Leukemia. EBioMedicine 2016, 14, 55–64. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Hurren, R.; MacLean, N.; Gronda, M.; Jitkova, Y.; Sukhai, M.A.; Minden, M.D.; Schimmer, A.D. Carnitine transporter CT2 (SLC22A16) is over-expressed in acute myeloid leukemia (AML) and target knockdown reduces growth and viability of AML cells. Apoptosis 2015, 20, 1099–1108. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Mardis, E.R.; Wilson, R.K. Cancer genome sequencing: A review. Hum. Mol. Genet. 2009. [Google Scholar] [CrossRef]
- Paschka, P.; Schlenk, R.F.; Gaidzik, V.I.; Habdank, M.; Kronke, J.; Bullinger, L.; Spath, D.; Kayser, S.; Zucknick, M.; Gotze, K.; et al. IDH1 and IDH2 mutations are frequent genetic alterations in acute myeloid leukemia and confer adverse prognosis in cytogenetically normal acute myeloid leukemia with NPM1 mutation without FLT3 internal tandem duplication. J. Clin. Oncol. 2010, 28, 3636–3643. [Google Scholar] [CrossRef]
- Marcucci, G.; Maharry, K.; Wu, Y.Z.; Radmacher, M.D.; Mrozek, K.; Margeson, D.; Holland, K.B.; Whitman, S.P.; Becker, H.; Schwind, S.; et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: A Cancer and Leukemia Group B study. J. Clin. Oncol. 2010, 28, 2348–2355. [Google Scholar] [CrossRef]
- Patel, J.P.; Gonen, M.; Figueroa, M.E.; Fernandez, H.; Sun, Z.; Racevskis, J.; Van Vlierberghe, P.; Dolgalev, I.; Thomas, S.; Aminova, O.; et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N. Engl. J. Med. 2012, 366, 1079–1089. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef]
- Chou, W.C.; Hou, H.A.; Chen, C.Y.; Tang, J.L.; Yao, M.; Tsay, W.; Ko, B.S.; Wu, S.J.; Huang, S.Y.; Hsu, S.C.; et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood 2010, 115, 2749–2754. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.M.; Majeti, R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 648–657. [Google Scholar] [CrossRef]
- Ferret, Y.; Boissel, N.; Helevaut, N.; Madic, J.; Nibourel, O.; Marceau-Renaut, A.; Bucci, M.; Geffroy, S.; Celli-Lebras, K.; Castaigne, S.; et al. Clinical relevance of IDH1/2 mutant allele burden during follow-up in acute myeloid leukemia. A study by the French ALFA group. Haematologica 2018, 103, 822–829. [Google Scholar] [CrossRef]
- Xu, Q.; Li, Y.; Lv, N.; Jing, Y.; Xu, Y.; Li, Y.; Li, W.; Yao, Z.; Chen, X.; Huang, S.; et al. Correlation Between Isocitrate Dehydrogenase Gene Aberrations and Prognosis of Patients with Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis. Clin. Cancer Res. 2017, 23, 4511–4522. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Chapuis, N.; Tamburini, J.; Bardet, V.; Cornillet-Lefebvre, P.; Willems, L.; Green, A.; Mayeux, P.; Lacombe, C.; Bouscary, D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica 2010, 95, 819–828. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef] [Green Version]
- Horne, G.A.; Kinstrie, R.; Copland, M. Novel drug therapies in myeloid leukemia. Pharm Pat. Anal. 2015, 4, 187–205. [Google Scholar] [CrossRef]
- Tamburini, J.; Elie, C.; Bardet, V.; Chapuis, N.; Park, S.; Broet, P.; Cornillet-Lefebvre, P.; Lioure, B.; Ugo, V.; Blanchet, O.; et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood 2007, 110, 1025–1028. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Simpson, S.E.; Scialla, T.J.; Bagg, A.; Carroll, M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood 2003, 102, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Gurumurthy, S.; Xie, S.Z.; Alagesan, B.; Kim, J.; Yusuf, R.Z.; Saez, B.; Tzatsos, A.; Ozsolak, F.; Milos, P.; Ferrari, F.; et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature 2010, 468, 659–663. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, A.; Azevedo, M.M.; Pereira, O.; Sampaio-Marques, B.; Paiva, A.; Correia-Neves, M.; Castro, I.; Ludovico, P. Proteolytic systems and AMP-activated protein kinase are critical targets of acute myeloid leukemia therapeutic approaches. Oncotarget 2015, 6, 31428–31440. [Google Scholar] [CrossRef]
- Hirao, A.; Hoshii, T. Mechanistic / mammalian target protein of rapamycin signaling in hematopoietic stem cells and leukemia. Cancer Sci. 2013, 104, 977–982. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [Green Version]
- Visnjic, D.; Dembitz, V.; Lalic, H. The role of AMPK/mTOR modulators in therapy of acute myeloid leukemia. Curr Med. Chem 2018. [Google Scholar] [CrossRef]
- Saito, Y.; Chapple, R.H.; Lin, A.; Kitano, A.; Nakada, D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell 2015, 17, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Sujobert, P.; Poulain, L.; Paubelle, E.; Zylbersztejn, F.; Grenier, A.; Lambert, M.; Townsend, E.C.; Brusq, J.M.; Nicodeme, E.; Decrooqc, J.; et al. Co-activation of AMPK and mTORC1 Induces Cytotoxicity in Acute Myeloid Leukemia. Cell Rep. 2015, 11, 1446–1457. [Google Scholar] [CrossRef]
- Sujobert, P.; Tamburini, J. Co-activation of AMPK and mTORC1 as a new therapeutic option for acute myeloid leukemia. Mol. Cell Oncol 2016. [Google Scholar] [CrossRef]
- Green, A.S.; Chapuis, N.; Maciel, T.T.; Willems, L.; Lambert, M.; Arnoult, C.; Boyer, O.; Bardet, V.; Park, S.; Foretz, M.; et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood 2010, 116, 4262–4273. [Google Scholar] [CrossRef] [Green Version]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef]
- Recher, C.; Beyne-Rauzy, O.; Demur, C.; Chicanne, G.; Dos Santos, C.; Mas, V.M.; Benzaquen, D.; Laurent, G.; Huguet, F.; Payrastre, B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005, 105, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.E.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef]
- Kung, C.P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M. Autophagy in tumor suppression and cancer therapy. Crit Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar] [CrossRef]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1. [Google Scholar] [CrossRef]
- Jin, J.; Britschgi, A.; Schlafli, A.M.; Humbert, M.; Shan-Krauer, D.; Batliner, J.; Federzoni, E.A.; Ernst, M.; Torbett, B.E.; Yousefi, S.; et al. Low Autophagy (ATG) Gene Expression Is Associated with an Immature AML Blast Cell Phenotype and Can Be Restored during AML Differentiation Therapy. Oxid Med. Cell Longev. 2018. [Google Scholar] [CrossRef]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Buhler, C.; Marcantonio, D.D.; Martinez, E.; Gollner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2017, 31, 505–510. [Google Scholar] [CrossRef]
- Walter, M.J.; Payton, J.E.; Ries, R.E.; Shannon, W.D.; Deshmukh, H.; Zhao, Y.; Baty, J.; Heath, S.; Westervelt, P.; Watson, M.A.; et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc. Natl Acad Sci U S A 2009, 106, 12950–12955. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis 2017. [Google Scholar] [CrossRef]
- Liu, T.; Kishton, R.J.; Macintyre, A.N.; Gerriets, V.A.; Xiang, H.; Liu, X.; Abel, E.D.; Rizzieri, D.; Locasale, J.W.; Rathmell, J.C. Glucose transporter 1-mediated glucose uptake is limiting for B-cell acute lymphoblastic leukemia anabolic metabolism and resistance to apoptosis. Cell Death Dis 2014. [Google Scholar] [CrossRef]
- Dalva-Aydemir, S.; Bajpai, R.; Martinez, M.; Adekola, K.U.; Kandela, I.; Wei, C.; Singhal, S.; Koblinski, J.E.; Raje, N.S.; Rosen, S.T.; et al. Targeting the metabolic plasticity of multiple myeloma with FDA-approved ritonavir and metformin. Clin. Cancer Res. 2015, 21, 1161–1171. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Vergez, F.; Serhan, N.; Delabesse, E.; Mansat-De Mas, V.; Hospital, M.A.; Tamburini, J.; Manenti, S.; Sarry, J.E.; et al. Antileukemic Activity of 2-Deoxy-d-Glucose through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia with FLT3-ITD or c-KIT Mutations. Mol. Cancer Ther. 2015, 14, 2364–2373. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.; Ju, H.Q.; Liu, K.; Zhan, G.; Liu, D.; Wen, S.; Garcia-Manero, G.; Huang, P.; Hu, Y. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer Lett. 2016, 377, 149–157. [Google Scholar] [CrossRef]
- Yang, H.; Tabe, Y.; Sekihara, K.; Saito, K.; Ma, H.; Ruvolo, V.; Imoto, J.; Ikeo, K.; Mogushi, K.; Hosoya, M.; et al. Novel Oxidative Phosphorylation Inhibitor IACS-010759 Induces AMPK-Dependent Apoptosis of AML Cells. Blood 2017, 130, 1245. [Google Scholar]
- Jacque, N.; Ronchetti, A.M.; Larrue, C.; Meunier, G.; Birsen, R.; Willems, L.; Saland, E.; Decroocq, J.; Maciel, T.T.; Lambert, M.; et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood 2015, 126, 1346–1356. [Google Scholar] [CrossRef] [Green Version]
- Cai, T.; Lorenzi, P.L.; Rakheja, D.; Pontikos, M.A.; Lodi, A.; Han, L.; Zhang, Q.; Ma, H.; Rahmani, M.; Bhagat, T.D.; et al. Gls Inhibitor CB-839 Modulates Cellular Metabolism in AML and Potently Suppresses AML Cell Growth When Combined with 5-Azacitidine. Blood 2016, 128, 4064. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov 2019, 9, 370–383. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Li, J.; Song, P.; Zhu, L.; Aziz, N.; Zhou, Q.; Zhang, Y.; Xu, W.; Feng, L.; Chen, D.; Wang, X.; et al. Synthetic lethality of glutaminolysis inhibition, autophagy inactivation and asparagine depletion in colon cancer. Oncotarget 2017, 8, 42664–42672. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Reisz, J.A.; Zaberezhnyy, V.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp. Hematol. 2018, 58, 52–58. [Google Scholar] [CrossRef]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef]
- Luo, M.; Brooks, M.; Wicha, M.S. Asparagine and Glutamine: Co-conspirators Fueling Metastasis. Cell Metab. 2018, 27, 947–949. [Google Scholar] [CrossRef] [Green Version]
- Mussai, F.; Egan, S.; Higginbotham-Jones, J.; Perry, T.; Beggs, A.; Odintsova, E.; Loke, J.; Pratt, G.; U, K.P.; Lo, A.; et al. Arginine dependence of acute myeloid leukemia blast proliferation: A novel therapeutic target. Blood 2015, 125, 2386–2396. [Google Scholar] [CrossRef]
- Southam, A.D.; Khanim, F.L.; Hayden, R.E.; Constantinou, J.K.; Koczula, K.M.; Michell, R.H.; Viant, M.R.; Drayson, M.T.; Bunce, C.M. Drug Redeployment to Kill Leukemia and Lymphoma Cells by Disrupting SCD1-Mediated Synthesis of Monounsaturated Fatty Acids. Cancer Res. 2015, 75, 2530–2540. [Google Scholar] [CrossRef] [Green Version]
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foa, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929. [Google Scholar] [CrossRef]
- Flaveny, C.A.; Griffett, K.; El-Gendy Bel, D.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Samudio, I.; Konopleva, M. Targeting leukemia’s "fatty tooth". Blood 2015, 126, 1874–1875. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef]
- Dang, L.; Yen, K.; Attar, E.C. IDH mutations in cancer and progress toward development of targeted therapeutics. Ann. Oncol. 2016, 27, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Kats, L.M.; Reschke, M.; Taulli, R.; Pozdnyakova, O.; Burgess, K.; Bhargava, P.; Straley, K.; Karnik, R.; Meissner, A.; Small, D.; et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell 2014, 14, 329–341. [Google Scholar] [CrossRef]
- Yen, K.; Travins, J.; Wang, F.; David, M.D.; Artin, E.; Straley, K.; Padyana, A.; Gross, S.; DeLaBarre, B.; Tobin, E.; et al. AG-221, a First-in-Class Therapy Targeting Acute Myeloid Leukemia Harboring Oncogenic IDH2 Mutations. Cancer Discov. 2017, 7, 478–493. [Google Scholar] [CrossRef]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- Fathi, A.T.; DiNardo, C.D.; Kline, I.; Kenvin, L.; Gupta, I.; Attar, E.C.; Stein, E.M.; de Botton, S.; Investigators, A.C.S. Differentiation Syndrome Associated With Enasidenib, a Selective Inhibitor of Mutant Isocitrate Dehydrogenase 2: Analysis of a Phase 1/2 Study. JAMA Oncol. 2018, 4, 1106–1110. [Google Scholar] [CrossRef]
- Norsworthy, K.J.; Mulkey, F.; Ward, A.F.; Przepiorka, D.; Deisseroth, A.B.; Farrell, A.T.; Pazdur, R. Incidence of Differentiation Syndrome with Ivosidenib (IVO) and Enasidenib (ENA) for Treatment of Patients with Relapsed or Refractory (R/R) Isocitrate Dehydrogenase (IDH)1- or IDH2-Mutated Acute Myeloid Leukemia (AML): A Systematic Analysis By the U.S. Food and Drug Administration (FDA). Blood 2018, 132. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Stein, E.M.; de Botton, S.; Roboz, G.J.; Altman, J.K.; Mims, A.S.; Swords, R.; Collins, R.H.; Mannis, G.N.; Pollyea, D.A.; et al. Durable Remissions with Ivosidenib in IDH1-Mutated Relapsed or Refractory AML. N. Engl. J. Med. 2018, 378, 2386–2398. [Google Scholar] [CrossRef]
- Huls, G. Azacitidine in AML: A treatment option? Blood 2015, 126, 283–284. [Google Scholar] [CrossRef]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef]
- Welch, J.S.; Petti, A.A.; Miller, C.A.; Fronick, C.C.; O’Laughlin, M.; Fulton, R.S.; Wilson, R.K.; Baty, J.D.; Duncavage, E.J.; Tandon, B.; et al. TP53 and Decitabine in Acute Myeloid Leukemia and Myelodysplastic Syndromes. N. Engl. J. Med. 2016, 375, 2023–2036. [Google Scholar] [CrossRef]
- Wouters, B.J.; Delwel, R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood 2016, 127, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Issa, J.J.; Roboz, G.; Rizzieri, D.; Jabbour, E.; Stock, W.; O’Connell, C.; Yee, K.; Tibes, R.; Griffiths, E.A.; Walsh, K.; et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: A multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 2015, 16, 1099–1110. [Google Scholar] [CrossRef]
- Daver, N.; Kantarjian, H.; Ravandi, F.; Estey, E.; Wang, X.; Garcia-Manero, G.; Jabbour, E.; Konopleva, M.; O’Brien, S.; Verstovsek, S.; et al. A phase II study of decitabine and gemtuzumab ozogamicin in newly diagnosed and relapsed acute myeloid leukemia and high-risk myelodysplastic syndrome. Leukemia 2016, 30, 268–273. [Google Scholar] [CrossRef]
- Yogarajah, M.; Stone, R.M. A concise review of BCL-2 inhibition in acute myeloid leukemia. Expert Rev. Hematol 2018, 11, 145–154. [Google Scholar] [CrossRef]
- Wei, A.; Strickland, S.A.; Roboz, G.J.; Hou, J.-Z.; Fiedler, W.; Lin, T.L.; Martinelli, G.; Walter, R.B.; Enjeti, A.; Fakouhi, K.; et al. Safety and Efficacy of Venetoclax Plus Low-Dose Cytarabine in Treatment-Naive Patients Aged ≥65 Years with Acute Myeloid Leukemia. Blood 2016, 128, 102–103. [Google Scholar]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Chan, S.M.; Thomas, D.; Corces-Zimmerman, M.R.; Xavy, S.; Rastogi, S.; Hong, W.J.; Zhao, F.; Medeiros, B.C.; Tyvoll, D.A.; Majeti, R. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat. Med. 2015, 21, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Floisand, Y.; et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2017, 31, 301–309. [Google Scholar] [CrossRef]
- Zeng, Z.; Shi, Y.X.; Tsao, T.; Qiu, Y.; Kornblau, S.M.; Baggerly, K.A.; Liu, W.; Jessen, K.; Liu, Y.; Kantarjian, H.; et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood 2012, 120, 2679–2689. [Google Scholar] [CrossRef]
- Rizzieri, D.A.; Feldman, E.; Dipersio, J.F.; Gabrail, N.; Stock, W.; Strair, R.; Rivera, V.M.; Albitar, M.; Bedrosian, C.L.; Giles, F.J. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 2008, 14, 2756–2762. [Google Scholar] [CrossRef]
- Tabe, Y.; Tafuri, A.; Sekihara, K.; Yang, H.; Konopleva, M. Inhibition of mTOR kinase as a therapeutic target for acute myeloid leukemia. Expert Opin. Ther. Targets 2017, 21, 705–714. [Google Scholar] [CrossRef]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino acid signalling upstream of mTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, W.; Ding, Y.; Xiu, B.; Li, P.; Dong, Y.; Zhu, Q.; Liang, A. Up-regulation of VEGF and its receptor in refractory leukemia cells. Int J. Clin. Exp. Pathol. 2015, 8, 5282–5290. [Google Scholar]
- Nobrega-Pereira, S.; Caiado, F.; Carvalho, T.; Matias, I.; Graca, G.; Goncalves, L.G.; Silva-Santos, B.; Norell, H.; Dias, S. VEGFR2-Mediated Reprogramming of Mitochondrial Metabolism Regulates the Sensitivity of Acute Myeloid Leukemia to Chemotherapy. Cancer Res. 2018, 78, 731–741. [Google Scholar] [CrossRef]
- Martelli, A.M.; Chiarini, F.; Evangelisti, C.; Ognibene, A.; Bressanin, D.; Billi, A.M.; Manzoli, L.; Cappellini, A.; McCubrey, J.A. Targeting the liver kinase B1/AMP-activated protein kinase pathway as a therapeutic strategy for hematological malignancies. Expert Opin. Ther. Targets 2012, 16, 729–742. [Google Scholar] [CrossRef]
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef]
- Scotland, S.; Saland, E.; Skuli, N.; de Toni, F.; Boutzen, H.; Micklow, E.; Senegas, I.; Peyraud, R.; Peyriga, L.; Theodoro, F.; et al. Mitochondrial energetic and AKT status mediate metabolic effects and apoptosis of metformin in human leukemic cells. Leukemia 2013, 27, 2129–2138. [Google Scholar] [CrossRef]
- Dinner, S.; Platanias, L.C. Targeting the mTOR Pathway in Leukemia. J. Cell Biochem. 2016, 117, 1745–1752. [Google Scholar] [CrossRef]
- Herschbein, L.; Liesveld, J.L. Dueling for dual inhibition: Means to enhance effectiveness of PI3K/Akt/mTOR inhibitors in AML. Blood Rev. 2018, 32, 235–248. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Rothe, K.; Porter, V.; Jiang, X. Current Outlook on Autophagy in Human Leukemia: Foe in Cancer Stem Cells and Drug Resistance, Friend in New Therapeutic Interventions. Int J. Mol. Sci. 2019, 20, 461. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N., Jr.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Chen, L.; Atkinson, J.M.; Claxton, D.F.; Wang, H.G. Atg5-dependent autophagy contributes to the development of acute myeloid leukemia in an MLL-AF9-driven mouse model. Cell Death Dis. 2016. [Google Scholar] [CrossRef]
- Porter, A.H.; Leveque-El Mouttie, L.; Vu, T.; Bruedigam, C.; Sutton, J.; Jacquelin, S.; Hill, G.R.; MacDonald, K.P.A.; Lane, S.W. Acute myeloid leukemia stem cell function is preserved in the absence of autophagy. Haematologica 2017. [Google Scholar] [CrossRef]
- Chen, X.; Clark, J.; Wunderlich, M.; Fan, C.; Davis, A.; Chen, S.; Guan, J.L.; Mulloy, J.C.; Kumar, A.; Zheng, Y. Autophagy is dispensable for Kmt2a/Mll-Mllt3/Af9 AML maintenance and anti-leukemic effect of chloroquine. Autophagy 2017, 13, 955–966. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Bosnjak, M.; Ristic, B.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Perovic, V.; Bogdanovic, A.; Paunovic, V.; Markovic, I.; Bumbasirevic, V.; et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Piya, S.; Andreeff, M.; Borthakur, G. Targeting autophagy to overcome chemoresistance in acute myleogenous leukemia. Autophagy 2017, 13, 214–215. [Google Scholar] [CrossRef]
- Walter, R.B.; Othus, M.; Burnett, A.K.; Lowenberg, B.; Kantarjian, H.M.; Ossenkoppele, G.J.; Hills, R.K.; Ravandi, F.; Pabst, T.; Evans, A.; et al. Resistance prediction in AML: Analysis of 4601 patients from MRC/NCRI, HOVON/SAKK, SWOG and MD Anderson Cancer Center. Leukemia 2015, 29, 312–320. [Google Scholar] [CrossRef]
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Bene, M.C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S.; et al. Minimal/measurable residual disease in AML: A consensus document from the European LeukemiaNet MRD Working Party. Blood 2018, 131, 1275–1291. [Google Scholar] [CrossRef]
- Hauge, M.; Bruserud, O.; Hatfield, K.J. Targeting of cell metabolism in human acute myeloid leukemia--more than targeting of isocitrate dehydrogenase mutations and PI3K/AKT/mTOR signaling? Eur. J. Haematol. 2016, 96, 211–221. [Google Scholar] [CrossRef]


{kind=link}
| Drugs | Drugs in Combination | Clinical Trial | Status |
|---|---|---|---|
| Targeting Oxidative Phosphorylation (OXPHOS) | |||
| IACS-010759 | NCT02882321 Phase I study to dose escalation and dose expansion of the IACS-010759. | Active, recruiting [79] | |
| Targeting amino acid metabolism | |||
| CB-839 | Azacitidine | NCT02071927 Phase I/II study to evaluate the dependence of AML and acute promyelocytic leukemia on glutamine. | Completed, no results posted [80,81] |
| Erwinase (ASPARAGINASE) | NCT02283190 Phase I study to determine and analyze the effects of Erwinaze/Asparaginase on reducing serum glutamine levels. | Completed, no results posted | |
| GRASPA (L-asparaginase encapsulated in red blood cells) | Low-dose cytarabine | NCT01810705 Phase II study to evaluate the efficacy and tolerability of GRASPA plus low-dose cytarabine in treatment of AML patients over 65 years old, unfit for intensive chemotherapy. | Completed, no results posted |
| PEG-BCT-100 (PEGylated recombinant human arginase) | NCT02899286 Phase II, non-randomized, open-label study to evaluate the efficacy of single agent PEG-BCT-100 on arginine depletion in adult patients with relapsed/refractory AML. | Active, not recruiting | |
| ADI-PEG 20 (arginine deiminase formulated with polyethlene glycol) | NCT01910012 Phase II study to evaluate the response rate of arginine depletion by arginine deiminase. | Active, not recruiting | |
| Targeting metabolic enzymes and epigenetic modifiers | |||
| Enasidenib (AG-221) | NCT02577406 Phase III study comparing the efficacy and safety of AG-221 versus conventional care regimens in subjects 60 years or older with AML refractory to or relapsed after second- or third-line AML therapy and positive for IDH2 mutation. | Active, recruiting | |
| Ivosidenib (AG-120) | Azacitidine | NCT03173248 Phase III study to evaluate the efficacy and safety of AG-120 plus azacitidine in adult subjects with previously untreated IDH1m AML who are considered appropriate candidates for non-intensive therapy. | Active, recruiting |
| IDH305 | NCT02381886 Phase I study to estimate the maximum tolerated dose/recommended dose for expansion of IDH305 in patients with advanced malignancies that harbor IDH1 R132 mutations. | Active, not recruiting | |
| BAY-1436032 | NCT03127735 Phase II study to determine the maximum tolerated and/or recommended dose of BAY1436032, a mIDH1 inhibitor in patients with mIDH1-R132X advanced AML. | Completed, no results posted | |
| AG-881 | NCT02492737 Phase I study to evaluate the safety, pharmacokinetics, pharmacodynamics, and clinical activity of AG-881 in advanced hematologic malignancies that harbor an IDH1 and/or IDH2 mutation. | Completed, no results posted | |
| FT-2102 | Azacitidine or cytarabine | NCT02719574 Phase I/II study to evaluate the safety efficacy, pharmacokinetics and pharmacodynamics of FT-2102 as a single agent or in combination with azacitidine or cytarabine. | Active, recruiting |
| Guadecitabine | NCT02920008 Phase III study of the efficacy and safety of guadecitabine in adults with previously treated AML. | Active, not recruiting | |
| Oral azacitidine | Best-supportive care | NCT01757535 Phase III study to compare efficacy and safety of oral azacitidine plus best-supportive care versus best supportive care as maintenance therapy in subjects with AML in complete remission. | Active, not recruiting |
| Azacitidine | Nivolumab | NCT02397720 Phase II study to determine the side effects and the best dose of nivolumab and azacitidine with or without ipilimumab for treating patients with AML that have not responded to previous treatment or have returned after a period of improvement or are newly diagnosed. | Recruiting [82] |
| Signaling pathways and autophagy inhibition | |||
| BKM120 | NCT01396499 Phase I study to determine the highest tolerable dose and the safety of BKM120, a PI3K kinase inhibitor that can be given to patients with relapsed or refractory leukemia. | Completed, no results posted | |
| ONC201 | Low-dose cytarabine | NCT02392572 Phase I/II study to find the highest tolerable dose and safety of ONC201, an inhibitor of the serine/threonine protein kinase AKT and extracellular signal-regulated kinase (ERK), alone or in combination with low dose cytarabine in patients with relapsed or refractory AML, acute promyelocytic leukemia, or myelodysplastic syndromes. | Active, recruiting |
| Everolimus | MICE-regimen followed by consolidation therapy with idarubicin, cytarabine, and etoposide | NCT01154439 Phase I study to determine the maximum-tolerated dose of everolimus, a derivative of rapamycin that inhibits mTOR, in combination with standard remission-induction therapy comprising mitoxantrone hydrochloride, cytarabine, and etoposide (MICE-regimen) followed by consolidation therapy comprising idarubicin, cytarabine, and etoposide in older patients with newly diagnosed AML. | Active, not recruiting |
| Sirolimus | Azacitidine | NCT01869114 Phase II trial studies how well sirolimus and azacitidine works in treating patients with high-risk myelodysplastic syndrome or recurrent AML. | Recruiting |
| Hydroxychloroquine (HCQ) | Mitoxantrone and Etoposide | NCT02631252 Phase I study to find the safest and most effective dose of hydroxychloroquine, an autophagy inhibitor, when combined with the usual medication for AML, mitoxantrone, and etoposide. | Terminated, no results posted |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. https://doi.org/10.3390/cells8090967
Castro I, Sampaio-Marques B, Ludovico P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells. 2019; 8(9):967. https://doi.org/10.3390/cells8090967
Chicago/Turabian StyleCastro, Isabel, Belém Sampaio-Marques, and Paula Ludovico. 2019. "Targeting Metabolic Reprogramming in Acute Myeloid Leukemia" Cells 8, no. 9: 967. https://doi.org/10.3390/cells8090967
APA StyleCastro, I., Sampaio-Marques, B., & Ludovico, P. (2019). Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells, 8(9), 967. https://doi.org/10.3390/cells8090967