Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD

{kind=link}
{kind=link}
Abstract
:1. Introduction
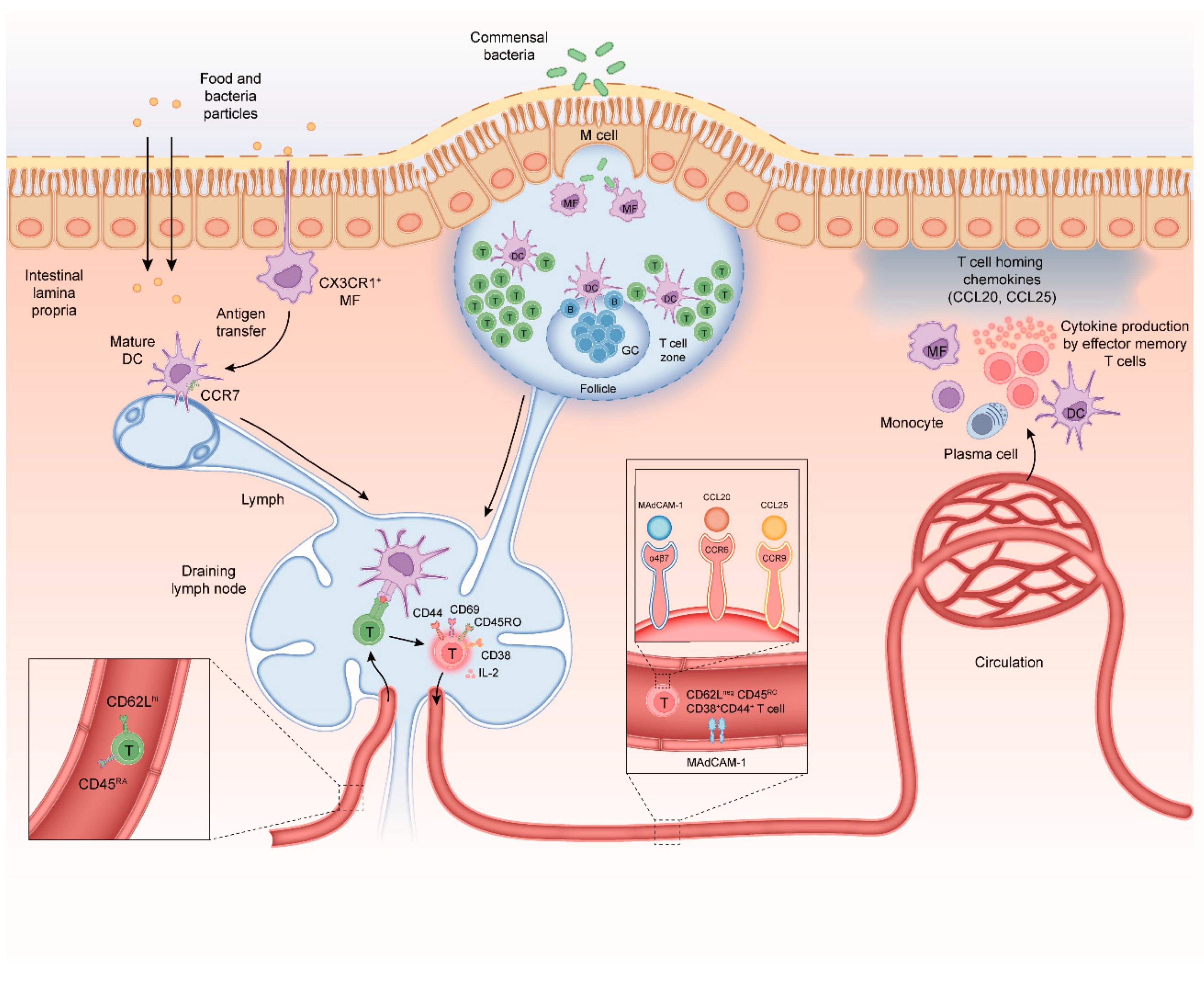
2. Mechanisms of CD4+ T-Cell Differentiation in the Healthy Intestine
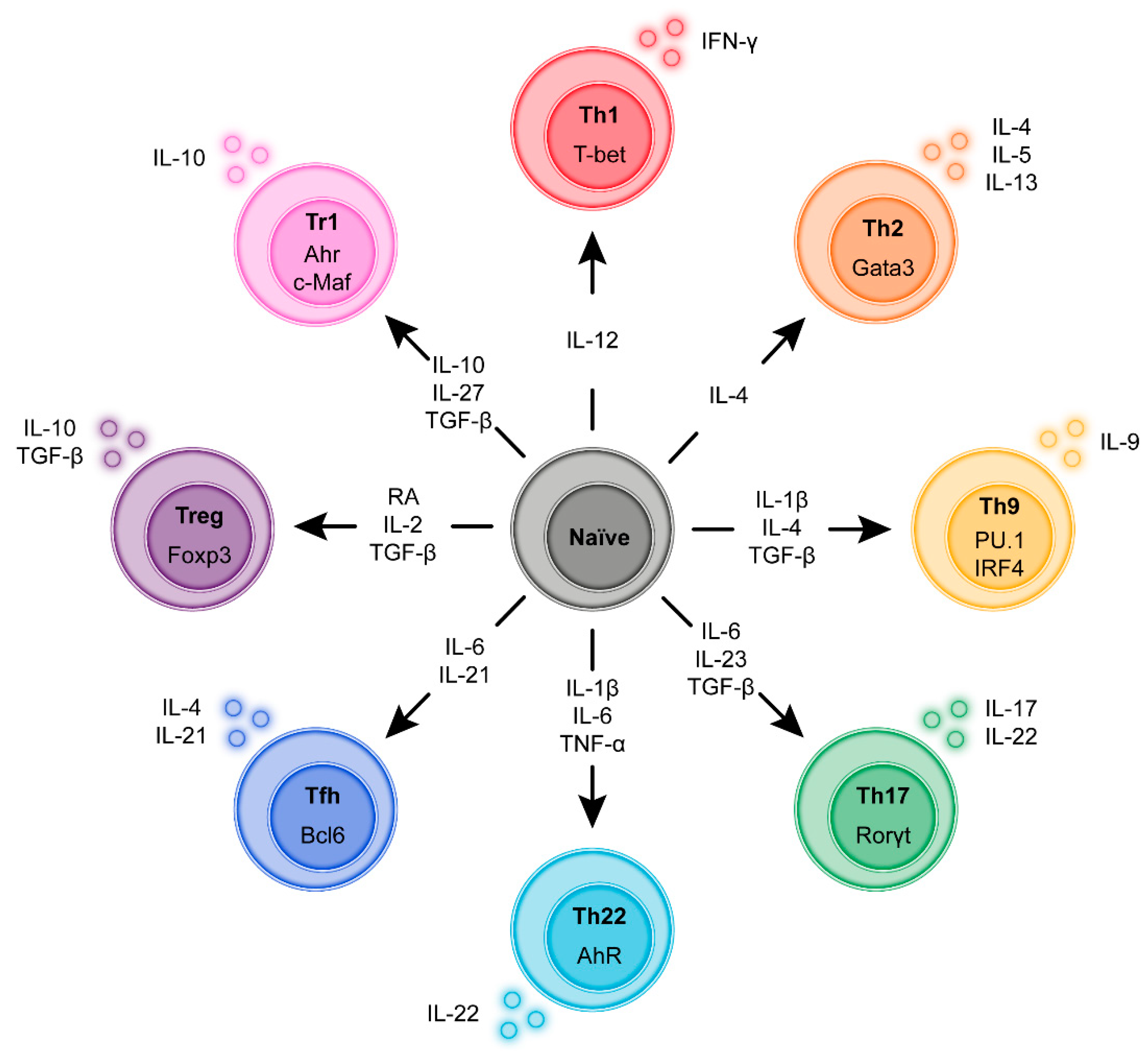
3. Regulatory and Inflammatory T-Cell Sub-Populations
3.1. T Helper 1 (Th1) Cells
3.2. T Helper 2 (Th2) Cells
3.3. T Helper 9 (Th9) Cells
3.4. T Helper 17 (Th17) Cells
3.5. T Helper 22 (Th22) Cells
3.6. T Follicular Helper (Tfh) Cells
3.7. T-Regulatory (Treg) Cells
3.8. Type 1 Regulatory T (Tr1) Cells
3.9. T-Cell Plasticity
4. T-Cells in IBD–What Are the Most Dominant Responses in Which Patients?
5. Abundance of IFN-γ Secreting CD4+ T-Cells in CD and UC
Does the Increased IFN-γ Response Contribute to Disease Pathology?
6. Abundance of IL-17 Secreting CD4+ T-Cells in CD and UC
6.1. Does the Increased IL-17 Response Contribute to Disease Pathology?
6.2. What Is the Cellular Source of IFN-γ and IL-17: Th1, Th17 or Th1/17?
7. The Role of IL-4, IL-5, and IL-13 Secreting CD4+ T-Cells in IBD
Does the Increased IL-13 Response Contribute to Disease Pathology?
8. The Role of IL-9 Secreting CD4+ T-Cells in IBD
Does the Increased IL-9 Response Contribute to Disease Pathology?
9. The Role of IL-22 Secreting CD4+ T-Cells in IBD
Does the IL-22–IL-22BP Pathway Contribute to Disease Pathology?
10. Regulatory T-Cell Populations (Treg and Tr1)
11. The Role of Tregs in IBD
Does Adoptive Transfer of Treg Attenuate Intestinal Disease?
12. The Role of Tr1 Cells in IBD
Does Adoptive Transfer of Tr1 Cells Attenuate Disease?
13. Toward Precision Intervention in Pathological T-Cell Responses in IBD
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Preza, G.C.; Yang, O.O.; Elliott, J.; Anton, P.A.; Ochoa, M.T. T lymphocyte density and distribution in human colorectal mucosa, and inefficiency of current cell isolation protocols. PLoS ONE 2015, 10, e0122723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowat, A.M. To respond or not to respond—A personal perspective of intestinal tolerance. Nat. Rev. Immunol. 2018, 18, 405–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; Meyer zum Buschenfelde, K.H. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 102, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.; Van Kemseke, C.; Reenaers, C. Necessity of phenotypic classification of inflammatory bowel disease. Best Pract Res. Clin. Gastroenterol. 2011, 25 (Suppl. 1), S2–S7. [Google Scholar] [CrossRef]
- Levine, A.; Griffiths, A.; Markowitz, J.; Wilson, D.C.; Turner, D.; Russell, R.K.; Fell, J.; Ruemmele, F.M.; Walters, T.; Sherlock, M.; et al. Pediatric Modification of the Montreal Classification for Inflammatory Bowel Disease: The Paris Classification. Inflamm. Bowel Dis. 2011, 17, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [Green Version]
- Dutton, R.W.; Bradley, L.M.; Swain, S.L. T cell memory. Annu. Rev. Immunol. 1998, 16, 201–223. [Google Scholar] [CrossRef]
- Emmrich, J.; Seyfarth, M.; Fleig, W.E.; Emmrich, F. Treatment of inflammatory bowel disease with anti-CD4 monoclonal antibody. Lancet 1991, 338, 570–571. [Google Scholar] [CrossRef]
- Stronkhorst, A.; Radema, S.; Yong, S.L.; Bijl, H.; ten Berge, I.J.; Tytgat, G.N.; van Deventer, S.J. CD4 antibody treatment in patients with active Crohn’s disease: A phase 1 dose finding study. Gut 1997, 40, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Shale, M.; Schiering, C.; Powrie, F. CD4(+) T-cell subsets in intestinal inflammation. Immunol. Rev. 2013, 252, 164–182. [Google Scholar] [CrossRef] [Green Version]
- Agace, W.W.; McCoy, K.D. Regionalized Development and Maintenance of the Intestinal Adaptive Immune Landscape. Immunity 2017, 46, 532–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Donaldson, D.S.; Ohno, H.; Williams, I.R.; Mahajan, A. Microfold (M) cells: Important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol. 2013, 6, 666–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niess, J.H.; Brand, S.; Gu, X.; Landsman, L.; Jung, S.; McCormick, B.A.; Vyas, J.M.; Boes, M.; Ploegh, H.L.; Fox, J.G.; et al. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science 2005, 307, 254–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, O.; Jaensson, E.; Persson, E.K.; Liu, X.; Worbs, T.; Agace, W.W.; Pabst, O. Intestinal CD103+, but not CX3CR1+, antigen sampling cells migrate in lymph and serve classical dendritic cell functions. J. Exp. Med. 2009, 206, 3101–3114. [Google Scholar] [CrossRef] [PubMed]
- Anjuere, F.; Luci, C.; Lebens, M.; Rousseau, D.; Hervouet, C.; Milon, G.; Holmgren, J.; Ardavin, C.; Czerkinsky, C. In vivo adjuvant-induced mobilization and maturation of gut dendritic cells after oral administration of cholera toxin. J. Immunol. 2004, 173, 5103–5111. [Google Scholar] [CrossRef]
- Johansson-Lindbom, B.; Svensson, M.; Pabst, O.; Palmqvist, C.; Marquez, G.; Forster, R.; Agace, W.W. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J. Exp. Med. 2005, 202, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Veenbergen, S.; van Berkel, L.A.; du Pre, M.F.; He, J.; Karrich, J.J.; Costes, L.M.; Luk, F.; Simons-Oosterhuis, Y.; Raatgeep, H.C.; Cerovic, V.; et al. Colonic tolerance develops in the iliac lymph nodes and can be established independent of CD103(+) dendritic cells. Mucosal Immunol. 2016, 9, 894–906. [Google Scholar] [CrossRef]
- Scott, C.L.; Bain, C.C.; Wright, P.B.; Sichien, D.; Kotarsky, K.; Persson, E.K.; Luda, K.; Guilliams, M.; Lambrecht, B.N.; Agace, W.W.; et al. CCR2(+)CD103(-) intestinal dendritic cells develop from DC-committed precursors and induce interleukin-17 production by T cells. Mucosal Immunol. 2015, 8, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Mayer, J.U.; Demiri, M.; Agace, W.W.; MacDonald, A.S.; Svensson-Frej, M.; Milling, S.W. Different populations of CD11b(+) dendritic cells drive Th2 responses in the small intestine and colon. Nat. Commun. 2017, 8, 15820. [Google Scholar] [CrossRef]
- Warnock, R.A.; Askari, S.; Butcher, E.C.; von Andrian, U.H. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J. Exp. Med. 1998, 187, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Catron, D.M.; Itano, A.A.; Pape, K.A.; Mueller, D.L.; Jenkins, M.K. Visualizing the first 50 hr of the primary immune response to a soluble antigen. Immunity 2004, 21, 341–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mempel, T.R.; Henrickson, S.E.; Von Andrian, U.H. T-cell priming by dendritic cells in lymph nodes occurs in three distinct phases. Nature 2004, 427, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K. The second touch hypothesis: T cell activation, homing and polarization. F1000Res 2014, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Du Pre, M.F.; van Berkel, L.A.; Raki, M.; van Leeuwen, M.A.; de Ruiter, L.F.; Broere, F.; Ter Borg, M.N.; Lund, F.E.; Escher, J.C.; Lundin, K.E.; et al. CD62L(neg)CD38(+) expression on circulating CD4(+) T cells identifies mucosally differentiated cells in protein fed mice and in human celiac disease patients and controls. Am. J. Gastroenterol. 2011, 106, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Joosse, M.E.; Menckeberg, C.L.; de Ruiter, L.F.; Raatgeep, H.R.C.; van Berkel, L.A.; Simons-Oosterhuis, Y.; Tindemans, I.; Muskens, A.F.M.; Hendriks, R.W.; Hoogenboezem, R.M.; et al. Frequencies of circulating regulatory TIGIT(+)CD38(+) effector T cells correlate with the course of inflammatory bowel disease. Mucosal Immunol. 2019, 12, 154–163. [Google Scholar] [CrossRef]
- Hamann, A.; Andrew, D.P.; Jablonski-Westrich, D.; Holzmann, B.; Butcher, E.C. Role of alpha 4-integrins in lymphocyte homing to mucosal tissues in vivo. J. Immunol. 1994, 152, 3282–3293. [Google Scholar]
- Svensson, M.; Marsal, J.; Ericsson, A.; Carramolino, L.; Broden, T.; Marquez, G.; Agace, W.W. CCL25 mediates the localization of recently activated CD8alphabeta(+) lymphocytes to the small-intestinal mucosa. J. Clin. Investig. 2002, 110, 1113–1121. [Google Scholar] [CrossRef]
- Iwata, M.; Hirakiyama, A.; Eshima, Y.; Kagechika, H.; Kato, C.; Song, S.Y. Retinoic acid imprints gut-homing specificity on T cells. Immunity 2004, 21, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Mora, J.R.; Bono, M.R.; Manjunath, N.; Weninger, W.; Cavanagh, L.L.; Rosemblatt, M.; Von Andrian, U.H. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 2003, 424, 88–93. [Google Scholar] [CrossRef]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Campbell, J.J.; Haraldsen, G.; Pan, J.; Boisvert, J.; Roberts, A.I.; Ebert, E.C.; Vierra, M.A.; Goodman, S.B.; Genovese, M.C.; et al. Lymphocyte CC chemokine receptor 9 and epithelial thymus-expressed chemokine (TECK) expression distinguish the small intestinal immune compartment: Epithelial expression of tissue-specific chemokines as an organizing principle in regional immunity. J. Exp. Med. 2000, 192, 761–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briskin, M.; Winsor-Hines, D.; Shyjan, A.; Cochran, N.; Bloom, S.; Wilson, J.; McEvoy, L.M.; Butcher, E.C.; Kassam, N.; Mackay, C.R.; et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am. J. Pathol. 1997, 151, 97–110. [Google Scholar]
- Habtezion, A.; Nguyen, L.P.; Hadeiba, H.; Butcher, E.C. Leukocyte Trafficking to the Small Intestine and Colon. Gastroenterology 2016, 150, 340–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadakis, K.A.; Prehn, J.; Nelson, V.; Cheng, L.; Binder, S.W.; Ponath, P.D.; Andrew, D.P.; Targan, S.R. The role of thymus-expressed chemokine and its receptor CCR9 on lymphocytes in the regional specialization of the mucosal immune system. J. Immunol. 2000, 165, 5069–5076. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hanly, E.K.; Wheeler, L.W.; Kaur, M.; McDonald, K.G.; Newberry, R.D. Effect of alpha4beta7 blockade on intestinal lymphocyte subsets and lymphoid tissue development. Inflamm. Bowel Dis. 2010, 16, 1751–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picarella, D.; Hurlbut, P.; Rottman, J.; Shi, X.; Butcher, E.; Ringler, D.J. Monoclonal antibodies specific for beta 7 integrin and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) reduce inflammation in the colon of scid mice reconstituted with CD45RBhigh CD4+ T cells. J. Immunol. 1997, 158, 2099–2106. [Google Scholar] [PubMed]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.P.; Pan, J.; Dinh, T.T.; Hadeiba, H.; O’Hara, E., 3rd; Ebtikar, A.; Hertweck, A.; Gokmen, M.R.; Lord, G.M.; Jenner, R.G.; et al. Role and species-specific expression of colon T cell homing receptor GPR15 in colitis. Nat. Immunol. 2015, 16, 207–213. [Google Scholar] [CrossRef]
- Oyama, T.; Miura, S.; Watanabe, C.; Hokari, R.; Fujiyama, Y.; Komoto, S.; Tsuzuki, Y.; Hosoe, N.; Nagata, H.; Hibi, T. CXCL12 and CCL20 play a significant role in mucosal T-lymphocyte adherence to intestinal microvessels in mice. Microcirculation 2007, 14, 753–766. [Google Scholar] [CrossRef]
- Targan, S.R.; Deem, R.L.; Liu, M.; Wang, S.; Nel, A. Definition of a lamina propria T cell responsive state. Enhanced cytokine responsiveness of T cells stimulated through the CD2 pathway. J. Immunol. 1995, 154, 664–675. [Google Scholar] [PubMed]
- De Maria, R.; Fais, S.; Silvestri, M.; Frati, L.; Pallone, F.; Santoni, A.; Testi, R. Continuous in vivo activation and transient hyporesponsiveness to TcR/CD3 triggering of human gut lamina propria lymphocytes. Eur. J. Immunol. 1993, 23, 3104–3108. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- DuPage, M.; Bluestone, J.A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 2016, 16, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th1/Th2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Spolski, R.; Liao, W.; Leonard, W.J. Complex interactions of transcription factors in mediating cytokine biology in T cells. Immunol. Rev. 2014, 261, 141–156. [Google Scholar] [CrossRef] [Green Version]
- Islam, S.A.; Luster, A.D. T cell homing to epithelial barriers in allergic disease. Nat. Med. 2012, 18, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, V.; Glimcher, L.H.; Lord, G.M. T-bet: A bridge between innate and adaptive immunity. Nat. Rev. Immunol. 2013, 13, 777–789. [Google Scholar] [CrossRef]
- Caza, T.; Landas, S. Functional and Phenotypic Plasticity of CD4(+) T Cell Subsets. Biomed. Res. Int. 2015, 2015, 521957. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.P. ‘All things considered’: Transcriptional regulation of T helper type 2 cell differentiation from precursor to effector activation. Immunology 2013, 140, 31–38. [Google Scholar] [CrossRef]
- Zheng, W.; Flavell, R.A. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 1997, 89, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.H.; Cohn, L.; Ray, P.; Bottomly, K.; Ray, A. Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 1997, 272, 21597–21603. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, E.; Bopp, T. Discovery and initial characterization of Th9 cells: The early years. Semin. Immunopathol. 2017, 39, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Finotto, S. IL-9 signaling as key driver of chronic inflammation in mucosal immunity. Cytokine Growth Factor Rev. 2016, 29, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.C.; et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(-) effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, C.; Turner, J.E.; Van Snick, J.; Stockinger, B. The many lives of IL-9: A question of survival? Nat. Immunol. 2012, 13, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Uyttenhove, C.; van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef]
- Chang, H.C.; Sehra, S.; Goswami, R.; Yao, W.; Yu, Q.; Stritesky, G.L.; Jabeen, R.; McKinley, C.; Ahyi, A.N.; Han, L.; et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010, 11, 527–534. [Google Scholar] [CrossRef] [Green Version]
- Staudt, V.; Bothur, E.; Klein, M.; Lingnau, K.; Reuter, S.; Grebe, N.; Gerlitzki, B.; Hoffmann, M.; Ulges, A.; Taube, C.; et al. Interferon-regulatory factor 4 is essential for the developmental program of T helper 9 cells. Immunity 2010, 33, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Xue, G.; Jin, G.; Fang, J.; Lu, Y. IL-4 together with IL-1beta induces antitumor Th9 cell differentiation in the absence of TGF-beta signaling. Nat. Commun. 2019, 10, 1376. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [Green Version]
- Mathur, A.N.; Chang, H.C.; Zisoulis, D.G.; Stritesky, G.L.; Yu, Q.; O’Malley, J.T.; Kapur, R.; Levy, D.E.; Kansas, G.S.; Kaplan, M.H. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 2007, 178, 4901–4907. [Google Scholar] [CrossRef]
- Hegazy, A.N.; West, N.R.; Stubbington, M.J.T.; Wendt, E.; Suijker, K.I.M.; Datsi, A.; This, S.; Danne, C.; Campion, S.; Duncan, S.H.; et al. Circulating and Tissue-Resident CD4(+) T Cells With Reactivity to Intestinal Microbiota Are Abundant in Healthy Individuals and Function Is Altered During Inflammation. Gastroenterology 2017, 153, 1320–1337. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Dai, D.; He, X.; Zhu, S.; Yao, Y.; Gao, H.; Wang, J.; Qu, F.; Qiu, J.; Wang, H.; et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 2015, 43, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Sundrud, M.S.; Skepner, J.; Yamagata, T. Targeting Th17 cells in autoimmune diseases. Trends Pharmacol. Sci. 2014, 35, 493–500. [Google Scholar] [CrossRef]
- Kinugasa, T.; Sakaguchi, T.; Gu, X.; Reinecker, H.C. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology 2000, 118, 1001–1011. [Google Scholar] [CrossRef]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duhen, T.; Geiger, R.; Jarrossay, D.; Lanzavecchia, A.; Sallusto, F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat. Immunol. 2009, 10, 857–863. [Google Scholar] [CrossRef]
- Plank, M.W.; Kaiko, G.E.; Maltby, S.; Weaver, J.; Tay, H.L.; Shen, W.; Wilson, M.S.; Durum, S.K.; Foster, P.S. Th22 Cells Form a Distinct Th Lineage from Th17 Cells In Vitro with Unique Transcriptional Properties and Tbet-Dependent Th1 Plasticity. J. Immunol. 2017, 198, 2182–2190. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Tsai, L.M.; Leong, Y.A.; Hu, X.; Ma, C.S.; Chevalier, N.; Sun, X.; Vandenberg, K.; Rockman, S.; Ding, Y.; et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity 2013, 39, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.K.; Rigby, R.J.; Zotos, D.; Tsai, L.M.; Kawamoto, S.; Marshall, J.L.; Ramiscal, R.R.; Chan, T.D.; Gatto, D.; Brink, R.; et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J. Exp. Med. 2011, 208, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Yan, X.; Zhong, B.; Nurieva, R.I.; Wang, A.; Wang, X.; Martin-Orozco, N.; Wang, Y.; Chang, S.H.; Esplugues, E.; et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J. Exp. Med. 2012, 209, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Hatzi, K.; Nance, J.P.; Kroenke, M.A.; Bothwell, M.; Haddad, E.K.; Melnick, A.; Crotty, S. BCL6 orchestrates Tfh cell differentiation via multiple distinct mechanisms. J. Exp. Med. 2015, 212, 539–553. [Google Scholar] [CrossRef] [Green Version]
- Moser, B. CXCR5, the Defining Marker for Follicular B Helper T (TFH) Cells. Front. Immunol. 2015, 6, 296. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.I.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.H.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthje, K.; Kallies, A.; Shimohakamada, Y.; Belz, G.T.; Light, A.; Tarlinton, D.M.; Nutt, S.L. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat. Immunol. 2012, 13, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Maceiras, A.R.; Fonseca, V.R.; Agua-Doce, A.; Graca, L. T follicular regulatory cells in mice and men. Immunology 2017, 152, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josefowicz, S.Z.; Rudensky, A. Control of regulatory T cell lineage commitment and maintenance. Immunity 2009, 30, 616–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [Green Version]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Itoh, M.; Takahashi, T.; Sakaguchi, N.; Kuniyasu, Y.; Shimizu, J.; Otsuka, F.; Sakaguchi, S. Thymus and autoimmunity: Production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J. Immunol. 1999, 162, 5317–5326. [Google Scholar]
- Hauet-Broere, F.; Unger, W.W.; Garssen, J.; Hoijer, M.A.; Kraal, G.; Samsom, J.N. Functional CD25- and CD25+ mucosal regulatory T cells are induced in gut-draining lymphoid tissue within 48 h after oral antigen application. Eur. J. Immunol. 2003, 33, 2801–2810. [Google Scholar] [CrossRef]
- Worbs, T.; Bode, U.; Yan, S.; Hoffmann, M.W.; Hintzen, G.; Bernhardt, G.; Forster, R.; Pabst, O. Oral tolerance originates in the intestinal immune system and relies on antigen carriage by dendritic cells. J. Exp. Med. 2006, 203, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenbergen, S.; Samsom, J.N. Maintenance of small intestinal and colonic tolerance by IL-10-producing regulatory T cell subsets. Curr. Opin. Immunol. 2012, 24, 269–276. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.S.; DiPaolo, R.J.; Andersson, J.; Shevach, E.M. Cutting Edge: IL-2 is essential for TGF-beta-mediated induction of Foxp3+ T regulatory cells. J. Immunol. 2007, 178, 4022–4026. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ioan-Facsinay, A.; van der Voort, E.I.; Huizinga, T.W.; Toes, R.E. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur. J. Immunol. 2007, 37, 129–138. [Google Scholar] [CrossRef]
- Mucida, D.; Park, Y.; Kim, G.; Turovskaya, O.; Scott, I.; Kronenberg, M.; Cheroutre, H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 2007, 317, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Coombes, J.L.; Siddiqui, K.R.; Arancibia-Carcamo, C.V.; Hall, J.; Sun, C.M.; Belkaid, Y.; Powrie, F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J. Exp. Med. 2007, 204, 1757–1764. [Google Scholar] [CrossRef]
- Sun, C.M.; Hall, J.A.; Blank, R.B.; Bouladoux, N.; Oukka, M.; Mora, J.R.; Belkaid, Y. Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J. Exp. Med. 2007, 204, 1775–1785. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [Green Version]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Asseman, C.; Mauze, S.; Leach, M.W.; Coffman, R.L.; Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999, 190, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Billmeier, U.; McHedlidze, T.; Blumberg, R.S.; Neurath, M.F. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology 2011, 141, 1875–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powrie, F.; Carlino, J.; Leach, M.W.; Mauze, S.; Coffman, R.L. A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med. 1996, 183, 2669–2674. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, S.; Malmstrom, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Read, S.; Greenwald, R.; Izcue, A.; Robinson, N.; Mandelbrot, D.; Francisco, L.; Sharpe, A.H.; Powrie, F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 2006, 177, 4376–4383. [Google Scholar] [CrossRef] [Green Version]
- Bacchetta, R.; Bigler, M.; Touraine, J.L.; Parkman, R.; Tovo, P.A.; Abrams, J.; de Waal Malefyt, R.; de Vries, J.E.; Roncarolo, M.G. High levels of interleukin 10 production in vivo are associated with tolerance in SCID patients transplanted with HLA mismatched hematopoietic stem cells. J. Exp. Med. 1994, 179, 493–502. [Google Scholar] [CrossRef]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M.; Gagliani, N. The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity 2018, 49, 1004–1019. [Google Scholar] [CrossRef] [Green Version]
- Bollyky, P.L.; Wu, R.P.; Falk, B.A.; Lord, J.D.; Long, S.A.; Preisinger, A.; Teng, B.; Holt, G.E.; Standifer, N.E.; Braun, K.R.; et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc. Natl. Acad. Sci. USA 2011, 108, 7938–7943. [Google Scholar] [CrossRef] [Green Version]
- Cobbold, S.P.; Nolan, K.F.; Graca, L.; Castejon, R.; Le Moine, A.; Frewin, M.; Humm, S.; Adams, E.; Thompson, S.; Zelenika, D.; et al. Regulatory T cells and dendritic cells in transplantation tolerance: Molecular markers and mechanisms. Immunol. Rev. 2003, 196, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Groux, H.; O’Garra, A.; Bigler, M.; Rouleau, M.; Antonenko, S.; de Vries, J.E.; Roncarolo, M.G. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 1997, 389, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Vieira, P.L.; Christensen, J.R.; Minaee, S.; O’Neill, E.J.; Barrat, F.J.; Boonstra, A.; Barthlott, T.; Stockinger, B.; Wraith, D.C.; O’Garra, A. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J. Immunol. 2004, 172, 5986–5993. [Google Scholar] [CrossRef] [Green Version]
- Huber, S.; Gagliani, N.; Esplugues, E.; O’Connor, W., Jr.; Huber, F.J.; Chaudhry, A.; Kamanaka, M.; Kobayashi, Y.; Booth, C.J.; Rudensky, A.Y.; et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3(-) and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 2011, 34, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlig, H.H.; Coombes, J.; Mottet, C.; Izcue, A.; Thompson, C.; Fanger, A.; Tannapfel, A.; Fontenot, J.D.; Ramsdell, F.; Powrie, F. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006, 177, 5852–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamanaka, M.; Kim, S.T.; Wan, Y.Y.; Sutterwala, F.S.; Lara-Tejero, M.; Galan, J.E.; Harhaj, E.; Flavell, R.A. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knockin tiger mouse. Immunity 2006, 25, 941–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, C.L.; Harrington, L.E.; Janowski, K.M.; Oliver, J.R.; Zindl, C.L.; Rudensky, A.Y.; Weaver, C.T. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3- precursor cells in the absence of interleukin 10. Nat. Immunol. 2007, 8, 931–941. [Google Scholar] [CrossRef]
- Glocker, E.O.; Kotlarz, D.; Boztug, K.; Gertz, E.M.; Schaffer, A.A.; Noyan, F.; Perro, M.; Diestelhorst, J.; Allroth, A.; Murugan, D.; et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 2009, 361, 2033–2045. [Google Scholar] [CrossRef] [Green Version]
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–1007 e1003. [Google Scholar] [CrossRef] [Green Version]
- Shouval, D.S.; Ouahed, J.; Biswas, A.; Goettel, J.A.; Horwitz, B.H.; Klein, C.; Muise, A.M.; Snapper, S.B. Interleukin 10 receptor signaling: Master regulator of intestinal mucosal homeostasis in mice and humans. Adv. Immunol. 2014, 122, 177–210. [Google Scholar] [CrossRef] [Green Version]
- Veenbergen, S.; van Leeuwen, M.A.; Driessen, G.J.; Kersseboom, R.; de Ruiter, L.F.; Raatgeep, R.H.C.; Lindenbergh-Kortleve, D.J.; Simons-Oosterhuis, Y.; Biermann, K.; Halley, D.J.J.; et al. Development and Function of Immune Cells in an Adolescent Patient With a Deficiency in the Interleukin-10 Receptor. J. Pediatr. Gastroenterol. Nutr. 2017, 65, e5–e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Shea, J.J.; Paul, W.E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 2010, 327, 1098–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Crome, S.Q.; MacDonald, K.G.; Dai, E.L.; Mager, D.L.; Levings, M.K. Human Th1 and Th17 cells exhibit epigenetic stability at signature cytokine and transcription factor loci. J. Immunol. 2011, 187, 5615–5626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15. [Google Scholar] [CrossRef]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Clemente-Casares, X.; Blanco, J.; Ambalavanan, P.; Yamanouchi, J.; Singha, S.; Fandos, C.; Tsai, S.; Wang, J.; Garabatos, N.; Izquierdo, C.; et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 2016, 530, 434–440. [Google Scholar] [CrossRef]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef]
- Pacciani, V.; Gregori, S.; Chini, L.; Corrente, S.; Chianca, M.; Moschese, V.; Rossi, P.; Roncarolo, M.G.; Angelini, F. Induction of anergic allergen-specific suppressor T cells using tolerogenic dendritic cells derived from children with allergies to house dust mites. J. Allergy Clin. Immunol. 2010, 125, 727–736. [Google Scholar] [CrossRef]
- Heinemann, C.; Heink, S.; Petermann, F.; Vasanthakumar, A.; Rothhammer, V.; Doorduijn, E.; Mitsdoerffer, M.; Sie, C.; Prazeres da Costa, O.; Buch, T.; et al. IL-27 and IL-12 oppose pro-inflammatory IL-23 in CD4+ T cells by inducing Blimp1. Nat. Commun. 2014, 5, 3770. [Google Scholar] [CrossRef] [Green Version]
- Koenen, H.J.; Smeets, R.L.; Vink, P.M.; van Rijssen, E.; Boots, A.M.; Joosten, I. Human CD25highFoxp3pos regulatory T cells differentiate into IL-17-producing cells. Blood 2008, 112, 2340–2352. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Villar, M.; Baecher-Allan, C.M.; Hafler, D.A. Identification of T helper type 1-like, Foxp3+ regulatory T cells in human autoimmune disease. Nat. Med. 2011, 17, 673–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClymont, S.A.; Putnam, A.L.; Lee, M.R.; Esensten, J.H.; Liu, W.; Hulme, M.A.; Hoffmuller, U.; Baron, U.; Olek, S.; Bluestone, J.A.; et al. Plasticity of human regulatory T cells in healthy subjects and patients with type 1 diabetes. J. Immunol. 2011, 186, 3918–3926. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.T.; Ong, D.E.; Lim, F.S.; Teng, K.W.; McGovern, N.; Narayanan, S.; Ho, W.Q.; Cerny, D.; Tan, H.K.; Anicete, R.; et al. A High-Dimensional Atlas of Human T Cell Diversity Reveals Tissue-Specific Trafficking and Cytokine Signatures. Immunity 2016, 45, 442–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smids, C.; Horjus Talabur Horje, C.S.; Drylewicz, J.; Roosenboom, B.; Groenen, M.J.M.; van Koolwijk, E.; van Lochem, E.G.; Wahab, P.J. Intestinal T Cell Profiling in Inflammatory Bowel Disease: Linking T Cell Subsets to Disease Activity and Disease Course. J. Crohns Colitis 2018, 12, 465–475. [Google Scholar] [CrossRef] [PubMed]
- Rubin, S.J.S.; Bai, L.; Haileselassie, Y.; Garay, G.; Yun, C.; Becker, L.; Streett, S.E.; Sinha, S.R.; Habtezion, A. Mass cytometry reveals systemic and local immune signatures that distinguish inflammatory bowel diseases. Nat. Commun. 2019, 10, 2686. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Weaver, C.T. Intestinal effector T cells in health and disease. Immunity 2009, 31, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory lymphocytes and intestinal inflammation. Annu. Rev. Immunol. 2009, 27, 313–338. [Google Scholar] [CrossRef]
- Patey-Mariaud de Serre, N.; Canioni, D.; Ganousse, S.; Rieux-Laucat, F.; Goulet, O.; Ruemmele, F.; Brousse, N. Digestive histopathological presentation of IPEX syndrome. Mod. Pathol. 2009, 22, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ueno, A.; Fort Gasia, M.; Luider, J.; Wang, T.; Hirota, C.; Jijon, H.B.; Deane, M.; Tom, M.; Chan, R.; et al. Profiles of Lamina Propria T Helper Cell Subsets Discriminate Between Ulcerative Colitis and Crohn’s Disease. Inflamm. Bowel Dis. 2016, 22, 1779–1792. [Google Scholar] [CrossRef]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar]
- Saruta, M.; Yu, Q.T.; Avanesyan, A.; Fleshner, P.R.; Targan, S.R.; Papadakis, K.A. Phenotype and effector function of CC chemokine receptor 9-expressing lymphocytes in small intestinal Crohn’s disease. J. Immunol. 2007, 178, 3293–3300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biancheri, P.; Di Sabatino, A.; Ammoscato, F.; Facciotti, F.; Caprioli, F.; Curciarello, R.; Hoque, S.S.; Ghanbari, A.; Joe-Njoku, I.; Giuffrida, P.; et al. Absence of a role for interleukin-13 in inflammatory bowel disease. Eur. J. Immunol. 2014, 44, 370–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verdier, J.; Begue, B.; Cerf-Bensussan, N.; Ruemmele, F.M. Compartmentalized expression of Th1 and Th17 cytokines in pediatric inflammatory bowel diseases. Inflamm. Bowel Dis. 2012, 18, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F.; Weigmann, B.; Finotto, S.; Glickman, J.; Nieuwenhuis, E.; Iijima, H.; Mizoguchi, A.; Mizoguchi, E.; Mudter, J.; Galle, P.R.; et al. The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J. Exp. Med. 2002, 195, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrello, T.; Monteleone, G.; Cucchiara, S.; Monteleone, I.; Sebkova, L.; Doldo, P.; Luzza, F.; Pallone, F. Up-regulation of the IL-12 receptor beta 2 chain in Crohn’s disease. J. Immunol 2000, 165, 7234–7239. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Matsumoto, T.; Esaki, M.; Torisu, T.; Iida, M. Profiles of circulating cytokines in patients with Crohn’s disease under maintenance therapy with infliximab. J. Crohns Colitis 2012, 6, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Papadakis, K.A.; Prehn, J.; Moreno, S.T.; Cheng, L.; Kouroumalis, E.A.; Deem, R.; Breaverman, T.; Ponath, P.D.; Andrew, D.P.; Green, P.H.; et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn’s disease. Gastroenterology 2001, 121, 246–254. [Google Scholar] [CrossRef]
- Kobayashi, T.; Okamoto, S.; Hisamatsu, T.; Kamada, N.; Chinen, H.; Saito, R.; Kitazume, M.T.; Nakazawa, A.; Sugita, A.; Koganei, K.; et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut 2008, 57, 1682–1689. [Google Scholar] [CrossRef] [Green Version]
- Kugathasan, S.; Saubermann, L.J.; Smith, L.; Kou, D.; Itoh, J.; Binion, D.G.; Levine, A.D.; Blumberg, R.S.; Fiocchi, C. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut 2007, 56, 1696–1705. [Google Scholar] [CrossRef]
- Zorzi, F.; Monteleone, I.; Sarra, M.; Calabrese, E.; Marafini, I.; Cretella, M.; Sedda, S.; Biancone, L.; Pallone, F.; Monteleone, G. Distinct profiles of effector cytokines mark the different phases of Crohn’s disease. PLoS ONE 2013, 8, e54562. [Google Scholar] [CrossRef]
- Reinisch, W.; Hommes, D.W.; Van Assche, G.; Colombel, J.F.; Gendre, J.P.; Oldenburg, B.; Teml, A.; Geboes, K.; Ding, H.; Zhang, L.; et al. A dose escalating, placebo controlled, double blind, single dose and multidose, safety and tolerability study of fontolizumab, a humanised anti-interferon gamma antibody, in patients with moderate to severe Crohn’s disease. Gut 2006, 55, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Hommes, D.W.; Mikhajlova, T.L.; Stoinov, S.; Stimac, D.; Vucelic, B.; Lonovics, J.; Zakuciova, M.; D’Haens, G.; Van Assche, G.; Ba, S.; et al. Fontolizumab, a humanised anti-interferon gamma antibody, demonstrates safety and clinical activity in patients with moderate to severe Crohn’s disease. Gut 2006, 55, 1131–1137. [Google Scholar] [CrossRef]
- Reinisch, W.; de Villiers, W.; Bene, L.; Simon, L.; Racz, I.; Katz, S.; Altorjay, I.; Feagan, B.; Riff, D.; Bernstein, C.N.; et al. Fontolizumab in moderate to severe Crohn’s disease: A phase 2, randomized, double-blind, placebo-controlled, multiple-dose study. Inflamm. Bowel Dis. 2010, 16, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Fuss, I.J.; Becker, C.; Yang, Z.; Groden, C.; Hornung, R.L.; Heller, F.; Neurath, M.F.; Strober, W.; Mannon, P.J. Both IL-12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 2006, 12, 9–15. [Google Scholar] [CrossRef]
- Uchida, S.; Oiso, N.; Komeda, Y.; Kudo, M.; Kawada, A. Paradoxical ulcerative colitis during treatment with secukinumab for psoriasis. Eur. J. Dermatol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [Green Version]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Jiang, W.; Su, J.; Zhang, X.; Cheng, X.; Zhou, J.; Shi, R.; Zhang, H. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm. Res. 2014, 63, 943–950. [Google Scholar] [CrossRef]
- Rovedatti, L.; Kudo, T.; Biancheri, P.; Sarra, M.; Knowles, C.H.; Rampton, D.S.; Corazza, G.R.; Monteleone, G.; Di Sabatino, A.; Macdonald, T.T. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 2009, 58, 1629–1636. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s Disease and Ulcerative Colitis Show Unique Cytokine Profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, M.J.; Karns, R.; Vallance, J.E.; Bezold, R.; Waddell, A.; Collins, M.H.; Haberman, Y.; Minar, P.; Baldassano, R.N.; Hyams, J.S.; et al. Mucosal Expression of Type 2 and Type 17 Immune Response Genes Distinguishes Ulcerative Colitis From Colon-Only Crohn’s Disease in Treatment-Naive Pediatric Patients. Gastroenterology 2017, 152, 1345–1357 e1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschek, M.A.; Boniface, K.; Sadekova, S.; Grein, J.; Murphy, E.E.; Turner, S.P.; Raskin, L.; Desai, B.; Faubion, W.A.; de Waal Malefyt, R.; et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 2009, 206, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Iboshi, Y.; Nakamura, K.; Fukaura, K.; Iwasa, T.; Ogino, H.; Sumida, Y.; Ihara, E.; Akiho, H.; Harada, N.; Nakamuta, M. Increased IL-17A/IL-17F expression ratio represents the key mucosal T helper/regulatory cell-related gene signature paralleling disease activity in ulcerative colitis. J. Gastroenterol. 2017, 52, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Holtta, V.; Klemetti, P.; Sipponen, T.; Westerholm-Ormio, M.; Kociubinski, G.; Salo, H.; Rasanen, L.; Kolho, K.L.; Farkkila, M.; Savilahti, E.; et al. IL-23/IL-17 immunity as a hallmark of Crohn’s disease. Inflamm. Bowel Dis. 2008, 14, 1175–1184. [Google Scholar] [CrossRef]
- Yen, D.; Cheung, J.; Scheerens, H.; Poulet, F.; McClanahan, T.; McKenzie, B.; Kleinschek, M.A.; Owyang, A.; Mattson, J.; Blumenschein, W.; et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 2006, 116, 1310–1316. [Google Scholar] [CrossRef]
- Elson, C.O.; Cong, Y.; Weaver, C.T.; Schoeb, T.R.; McClanahan, T.K.; Fick, R.B.; Kastelein, R.A. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 2007, 132, 2359–2370. [Google Scholar] [CrossRef]
- Kullberg, M.C.; Jankovic, D.; Feng, C.G.; Hue, S.; Gorelick, P.L.; McKenzie, B.S.; Cua, D.J.; Powrie, F.; Cheever, A.W.; Maloy, K.J.; et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006, 203, 2485–2494. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, A.; Andoh, A.; Araki, Y.; Bamba, T.; Fujiyama, Y. Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin. Immunol. 2004, 110, 55–62. [Google Scholar] [CrossRef]
- Becker, C.; Dornhoff, H.; Neufert, C.; Fantini, M.C.; Wirtz, S.; Huebner, S.; Nikolaev, A.; Lehr, H.A.; Murphy, A.J.; Valenzuela, D.M.; et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 2006, 177, 2760–2764. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, W., Jr.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, D.; Jamaluddin, N.; Pisegna, J.; Padua, D. A Challenging Case of Severe Ulcerative Colitis following the Initiation of Secukinumab for Ankylosing Spondylitis. Case Rep. Gastrointest. Med. 2018, 2018, 9679287. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, S.; Colombel, J.F.; Feagan, B.G.; Reich, K.; Deodhar, A.A.; McInnes, I.B.; Porter, B.; Das Gupta, A.; Pricop, L.; Fox, T. Incidence rates of inflammatory bowel disease in patients with psoriasis, psoriatic arthritis and ankylosing spondylitis treated with secukinumab: A retrospective analysis of pooled data from 21 clinical trials. Ann. Rheum. Dis. 2019, 78, 473–479. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.; Panaccione, R.; Khanna, R.; Feagan, B.G.; Jairath, V. IL12/23 or selective IL23 inhibition for the management of moderate-to-severe Crohn’s disease? Best Pract. Res. Clin. Gastroenterol. 2019, 38–39, 101604. [Google Scholar] [CrossRef]
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Fili, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861. [Google Scholar] [CrossRef] [Green Version]
- Globig, A.M.; Hennecke, N.; Martin, B.; Seidl, M.; Ruf, G.; Hasselblatt, P.; Thimme, R.; Bengsch, B. Comprehensive intestinal T helper cell profiling reveals specific accumulation of IFN-gamma+IL-17+coproducing CD4+ T cells in active inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 2321–2329. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Rott, L.; Kunkel, E.J.; Genovese, M.C.; Andrew, D.P.; Wu, L.; Butcher, E.C. Rules of chemokine receptor association with T cell polarization in vivo. J. Clin. Investig. 2001, 108, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [Green Version]
- Kebir, H.; Ifergan, I.; Alvarez, J.I.; Bernard, M.; Poirier, J.; Arbour, N.; Duquette, P.; Prat, A. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann. Neurol. 2009, 66, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Zielinski, C.E.; Lanzavecchia, A. Human Th17 subsets. Eur. J. Immunol. 2012, 42, 2215–2220. [Google Scholar] [CrossRef] [PubMed]
- Nistala, K.; Adams, S.; Cambrook, H.; Ursu, S.; Olivito, B.; de Jager, W.; Evans, J.G.; Cimaz, R.; Bajaj-Elliott, M.; Wedderburn, L.R. Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. USA 2010, 107, 14751–14756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef]
- Wang, Y.; Godec, J.; Ben-Aissa, K.; Cui, K.; Zhao, K.; Pucsek, A.B.; Lee, Y.K.; Weaver, C.T.; Yagi, R.; Lazarevic, V. The transcription factors T-bet and Runx are required for the ontogeny of pathogenic interferon-gamma-producing T helper 17 cells. Immunity 2014, 40, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Lubberts, E. The IL-23-IL-17 axis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 415–429. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef] [Green Version]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Burgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Frey, M.R.; Washington, M.K.; Chaturvedi, R.; Kuhnhein, L.A.; Matta, P.; Revetta, F.L.; Wilson, K.T.; Polk, D.B. STAT6 activation in ulcerative colitis: A new target for prevention of IL-13-induced colon epithelial cell dysfunction. Inflamm. Bowel Dis. 2011, 17, 2224–2234. [Google Scholar] [CrossRef]
- Popp, V.; Gerlach, K.; Mott, S.; Turowska, A.; Garn, H.; Atreya, R.; Lehr, H.A.; Ho, I.C.; Renz, H.; Weigmann, B.; et al. Rectal Delivery of a DNAzyme That Specifically Blocks the Transcription Factor GATA3 and Reduces Colitis in Mice. Gastroenterology 2017, 152, 176–192 e175. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Ohtsuka, Y.; Ikuse, T.; Baba, Y.; Yamakawa, Y.; Aoyagi, Y.; Fujii, T.; Kudo, T.; Nagata, S.; Shimizu, T. Increased mucosal expression of GATA-3 and STAT-4 in pediatric ulcerative colitis. Pediatr. Int. 2010, 52, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Heller, F.; Fuss, I.J.; Nieuwenhuis, E.E.; Blumberg, R.S.; Strober, W. Oxazolone colitis, a Th2 colitis model resembling ulcerative colitis, is mediated by IL-13-producing NK-T cells. Immunity 2002, 17, 629–638. [Google Scholar] [CrossRef] [Green Version]
- Reinisch, W.; Panes, J.; Khurana, S.; Toth, G.; Hua, F.; Comer, G.M.; Hinz, M.; Page, K.; O’Toole, M.; Moorehead, T.M.; et al. Anrukinzumab, an anti-interleukin 13 monoclonal antibody, in active UC: Efficacy and safety from a phase IIa randomised multicentre study. Gut 2015, 64, 894–900. [Google Scholar] [CrossRef]
- Danese, S.; Rudzinski, J.; Brandt, W.; Dupas, J.L.; Peyrin-Biroulet, L.; Bouhnik, Y.; Kleczkowski, D.; Uebel, P.; Lukas, M.; Knutsson, M.; et al. Tralokinumab for moderate-to-severe UC: A randomised, double-blind, placebo-controlled, phase IIa study. Gut 2015, 64, 243–249. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, P.; Caprioli, F.; Facciotti, F.; Di Sabatino, A. The role of interleukin-13 in chronic inflammatory intestinal disorders. Autoimmunity Rev. 2019, 18, 549–555. [Google Scholar] [CrossRef]
- Bailey, J.R.; Bland, P.W.; Tarlton, J.F.; Peters, I.; Moorghen, M.; Sylvester, P.A.; Probert, C.S.; Whiting, C.V. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: A role for innate lymphoid cells? PLoS ONE 2012, 7, e52332. [Google Scholar] [CrossRef] [Green Version]
- Curciarello, R.; Docena, G.H.; MacDonald, T.T. The Role of Cytokines in the Fibrotic Responses in Crohn’s Disease. Front. Med. 2017, 4, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steenwinckel, V.; Louahed, J.; Lemaire, M.M.; Sommereyns, C.; Warnier, G.; McKenzie, A.; Brombacher, F.; Van Snick, J.; Renauld, J.C. IL-9 promotes IL-13-dependent paneth cell hyperplasia and up-regulation of innate immunity mediators in intestinal mucosa. J. Immunol. 2009, 182, 4737–4743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalleweg, N.; Chiriac, M.T.; Podstawa, E.; Lehmann, C.; Rau, T.T.; Atreya, R.; Krauss, E.; Hundorfean, G.; Fichtner-Feigl, S.; Hartmann, A.; et al. IL-9 and its receptor are predominantly involved in the pathogenesis of UC. Gut 2015, 64, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlach, K.; Hwang, Y.; Nikolaev, A.; Atreya, R.; Dornhoff, H.; Steiner, S.; Lehr, H.A.; Wirtz, S.; Vieth, M.; Waisman, A.; et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat. Immunol. 2014, 15, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Matusiewicz, M.; Neubauer, K.; Bednarz-Misa, I.; Gorska, S.; Krzystek-Korpacka, M. Systemic interleukin-9 in inflammatory bowel disease: Association with mucosal healing in ulcerative colitis. World J. Gastroenterol. 2017, 23, 4039–4046. [Google Scholar] [CrossRef]
- Shohan, M.; Sabzevary-Ghahfarokhi, M.; Bagheri, N.; Shirzad, H.; Rahimian, G.; Soltani, A.; Ghatreh-Samani, M.; Deris, F.; Tahmasbi, K.; Shahverdi, E.; et al. Intensified Th9 Response is Associated with the Immunopathogenesis of Active Ulcerative Colitis. Immunol. Investig. 2018, 47, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, K.; McKenzie, A.N.; Neurath, M.F.; Weigmann, B. IL-9 regulates intestinal barrier function in experimental T cell-mediated colitis. Tissue Barriers 2015, 3, e983777. [Google Scholar] [CrossRef] [Green Version]
- Boland, B.S.; Sandborn, W.J.; Chang, J.T. Update on Janus kinase antagonists in inflammatory bowel disease. Gastroenterol. Clin. N. Am. 2014, 43, 603–617. [Google Scholar] [CrossRef] [Green Version]
- Kamanaka, M.; Huber, S.; Zenewicz, L.A.; Gagliani, N.; Rathinam, C.; O’Connor, W., Jr.; Wan, Y.Y.; Nakae, S.; Iwakura, Y.; Hao, L.; et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J. Exp. Med. 2011, 208, 1027–1040. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Izotova, L.S.; Mirochnitchenko, O.V.; Esterova, E.; Dickensheets, H.; Donnelly, R.P.; Pestka, S. Identification, cloning, and characterization of a novel soluble receptor that binds IL-22 and neutralizes its activity. J. Immunol. 2001, 166, 7096–7103. [Google Scholar] [CrossRef] [Green Version]
- Wolk, K.; Witte, E.; Hoffmann, U.; Doecke, W.D.; Endesfelder, S.; Asadullah, K.; Sterry, W.; Volk, H.D.; Wittig, B.M.; Sabat, R. IL-22 induces lipopolysaccharide-binding protein in hepatocytes: A potential systemic role of IL-22 in Crohn’s disease. J. Immunol. 2007, 178, 5973–5981. [Google Scholar] [CrossRef]
- Schmechel, S.; Konrad, A.; Diegelmann, J.; Glas, J.; Wetzke, M.; Paschos, E.; Lohse, P.; Goke, B.; Brand, S. Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL-22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm. Bowel Dis. 2008, 14, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Zhang, Z.; Inatomi, O.; Fujino, S.; Deguchi, Y.; Araki, Y.; Tsujikawa, T.; Kitoh, K.; Kim-Mitsuyama, S.; Takayanagi, A.; et al. Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005, 129, 969–984. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Li, J.; Ueno, A.; Iacucci, M.; Qian, J.; Ghosh, S. Histopathological Features of Inflammatory Bowel Disease are Associated With Different CD4+ T Cell Subsets in Colonic Mucosal Lamina Propria. J. Crohns Colitis 2018, 12, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Pang, Z.; Shu, W.; Wu, W.; Sun, M.; Cong, Y.; Liu, Z. Anti-TNF Therapy Induces CD4+ T-Cell Production of IL-22 and Promotes Epithelial Repairs in Patients With Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 1733–1744. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Esterhazy, D.; Canesso, M.C.C.; Mesin, L.; Muller, P.A.; de Castro, T.B.R.; Lockhart, A.; ElJalby, M.; Faria, A.M.C.; Mucida, D. Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nature 2019, 569, 126–130. [Google Scholar] [CrossRef]
- Houston, S.A.; Cerovic, V.; Thomson, C.; Brewer, J.; Mowat, A.M.; Milling, S. The lymph nodes draining the small intestine and colon are anatomically separate and immunologically distinct. Mucosal Immunol. 2016, 9, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R., Jr.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef]
- Santegoets, S.J.; Dijkgraaf, E.M.; Battaglia, A.; Beckhove, P.; Britten, C.M.; Gallimore, A.; Godkin, A.; Gouttefangeas, C.; de Gruijl, T.D.; Koenen, H.J.; et al. Monitoring regulatory T cells in clinical samples: Consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol. Immunother. 2015, 64, 1271–1286. [Google Scholar] [CrossRef] [Green Version]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Saruta, M.; Yu, Q.T.; Fleshner, P.R.; Mantel, P.Y.; Schmidt-Weber, C.B.; Banham, A.H.; Papadakis, K.A. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin. Immunol. 2007, 125, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.T.; Saruta, M.; Avanesyan, A.; Fleshner, P.R.; Banham, A.H.; Papadakis, K.A. Expression and functional characterization of FOXP3+ CD4+ regulatory T cells in ulcerative colitis. Inflamm. Bowel Dis. 2007, 13, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Holmen, N.; Lundgren, A.; Lundin, S.; Bergin, A.M.; Rudin, A.; Sjovall, H.; Ohman, L. Functional CD4+CD25high regulatory T cells are enriched in the colonic mucosa of patients with active ulcerative colitis and increase with disease activity. Inflamm. Bowel Dis. 2006, 12, 447–456. [Google Scholar] [CrossRef]
- Reikvam, D.H.; Perminow, G.; Lyckander, L.G.; Gran, J.M.; Brandtzaeg, P.; Vatn, M.; Carlsen, H.S. Increase of regulatory T cells in ileal mucosa of untreated pediatric Crohn’s disease patients. Scand. J. Gastroenterol. 2011, 46, 550–560. [Google Scholar] [CrossRef]
- Hovhannisyan, Z.; Treatman, J.; Littman, D.R.; Mayer, L. Characterization of interleukin-17-producing regulatory T cells in inflamed intestinal mucosa from patients with inflammatory bowel diseases. Gastroenterology 2011, 140, 957–965. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ueno, A.; Iacucci, M.; Fort Gasia, M.; Jijon, H.B.; Panaccione, R.; Kaplan, G.G.; Beck, P.L.; Luider, J.; Barkema, H.W.; et al. Crossover Subsets of CD4(+) T Lymphocytes in the Intestinal Lamina Propria of Patients with Crohn’s Disease and Ulcerative Colitis. Digestive Dis. Sci. 2017, 62, 2357–2368. [Google Scholar] [CrossRef]
- Ueno, A.; Jijon, H.; Chan, R.; Ford, K.; Hirota, C.; Kaplan, G.G.; Beck, P.L.; Iacucci, M.; Fort Gasia, M.; Barkema, H.W.; et al. Increased prevalence of circulating novel IL-17 secreting Foxp3 expressing CD4+ T cells and defective suppressive function of circulating Foxp3+ regulatory cells support plasticity between Th17 and regulatory T cells in inflammatory bowel disease patients. Inflamm. Bowel Dis. 2013, 19, 2522–2534. [Google Scholar] [CrossRef]
- Boden, E.K.; Lord, J.D. CD4 T Cells in IBD: Crossing the Line? Digestive Dis. Sci. 2017, 62, 2208–2210. [Google Scholar] [CrossRef] [Green Version]
- Ban, H.; Andoh, A.; Shioya, M.; Nishida, A.; Tsujikawa, T.; Fujiyama, Y. Increased number of FoxP3+CD4+ regulatory T cells in inflammatory bowel disease. Mol. Med. Rep. 2008, 1, 647–650. [Google Scholar] [CrossRef]
- Li, Z.; Vermeire, S.; Bullens, D.; Ferrante, M.; Van Steen, K.; Noman, M.; Rutgeerts, P.; Ceuppens, J.L.; Van Assche, G. Restoration of Foxp3+ Regulatory T-cell Subsets and Foxp3- Type 1 Regulatory-like T Cells in Inflammatory Bowel Diseases During Anti-tumor Necrosis Factor Therapy. Inflamm. Bowel Dis. 2015, 21, 2418–2428. [Google Scholar] [CrossRef] [Green Version]
- Veltkamp, C.; Anstaett, M.; Wahl, K.; Moller, S.; Gangl, S.; Bachmann, O.; Hardtke-Wolenski, M.; Langer, F.; Stremmel, W.; Manns, M.P.; et al. Apoptosis of regulatory T lymphocytes is increased in chronic inflammatory bowel disease and reversed by anti-TNFalpha treatment. Gut 2011, 60, 1345–1353. [Google Scholar] [CrossRef]
- Guidi, L.; Felice, C.; Procoli, A.; Bonanno, G.; Martinelli, E.; Marzo, M.; Mocci, G.; Pugliese, D.; Andrisani, G.; Danese, S.; et al. FOXP3(+) T regulatory cell modifications in inflammatory bowel disease patients treated with anti-TNFalpha agents. Biomed. Res. Int. 2013, 2013, 286368. [Google Scholar] [CrossRef] [Green Version]
- La Scaleia, R.; Morrone, S.; Stoppacciaro, A.; Scarpino, S.; Antonelli, M.; Bianchi, E.; Di Nardo, G.; Oliva, S.; Viola, F.; Cucchiara, S.; et al. Peripheral and intestinal CD4+ T cells with a regulatory phenotype in pediatric patients with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 563–572. [Google Scholar] [CrossRef]
- Li, Z.; Arijs, I.; De Hertogh, G.; Vermeire, S.; Noman, M.; Bullens, D.; Coorevits, L.; Sagaert, X.; Schuit, F.; Rutgeerts, P.; et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm. Bowel Dis. 2010, 16, 1299–1310. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Biancheri, P.; Piconese, S.; Rosado, M.M.; Ardizzone, S.; Rovedatti, L.; Ubezio, C.; Massari, A.; Sampietro, G.M.; Foschi, D.; et al. Peripheral regulatory T cells and serum transforming growth factor-beta: Relationship with clinical response to infliximab in Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 1891–1897. [Google Scholar] [CrossRef]
- Boschetti, G.; Nancey, S.; Sardi, F.; Roblin, X.; Flourie, B.; Kaiserlian, D. Therapy with anti-TNFalpha antibody enhances number and function of Foxp3(+) regulatory T cells in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 160–170. [Google Scholar] [CrossRef]
- Grundstrom, J.; Linton, L.; Thunberg, S.; Forsslund, H.; Janczewska, I.; Befrits, R.; van Hage, M.; Gafvelin, G.; Eberhardson, M. Altered immunoregulatory profile during anti-tumour necrosis factor treatment of patients with inflammatory bowel disease. Clin. Exp. Immunol. 2012, 169, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, R.; Scotta, C.; Cooper, D.; Nissim-Eliraz, E.; Nir, E.; Tasker, S.; Irving, P.M.; Sanderson, J.; Lavender, P.; Ibrahim, F.; et al. Correction of Defective T-Regulatory Cells From Patients With Crohn’s Disease by Ex Vivo Ligation of Retinoic Acid Receptor-alpha. Gastroenterology 2019, 156, 1775–1787. [Google Scholar] [CrossRef] [Green Version]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; Defor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef]
- Di Ianni, M.; Falzetti, F.; Carotti, A.; Terenzi, A.; Castellino, F.; Bonifacio, E.; Del Papa, B.; Zei, T.; Ostini, R.I.; Cecchini, D.; et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011, 117, 3921–3928. [Google Scholar] [CrossRef]
- Roncarolo, M.G.; Yssel, H.; Touraine, J.L.; Betuel, H.; De Vries, J.E.; Spits, H. Autoreactive T cell clones specific for class I and class II HLA antigens isolated from a human chimera. J. Exp. Med. 1988, 167, 1523–1534. [Google Scholar] [CrossRef] [Green Version]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Gonzalez, J.; Fuentes-Duculan, J.; Garcet, S.; Krueger, J.G. Proportion of CD4(+)CD49b(+)LAG-3(+) Type 1 Regulatory T Cells in the Blood of Psoriasis Patients Inversely Correlates with Psoriasis Area and Severity Index. J. Investig. Dermatol. 2018, 138, 2669–2672. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Vent-Schmidt, J.; McGeough, M.D.; Wong, M.; Hoffman, H.M.; Steiner, T.S.; Levings, M.K. Tr1 Cells, but Not Foxp3+ Regulatory T Cells, Suppress NLRP3 Inflammasome Activation via an IL-10-Dependent Mechanism. J. Immunol. 2015, 195, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [Green Version]
- Cook, L.; Stahl, M.; Han, X.; Nazli, A.; MacDonald, K.N.; Wong, M.Q.; Tsai, K.; Dizzell, S.; Jacobson, K.; Bressler, B.; et al. Suppressive and Gut Reparative Functions of Human Type 1 T-regulatory Cells. Gastroenterology 2019. [Google Scholar] [CrossRef] [Green Version]
- Alfen, J.S.; Larghi, P.; Facciotti, F.; Gagliani, N.; Bosotti, R.; Paroni, M.; Maglie, S.; Gruarin, P.; Vasco, C.M.; Ranzani, V.; et al. Intestinal IFN-gamma-producing type 1 regulatory T cells coexpress CCR5 and programmed cell death protein 1 and downregulate IL-10 in the inflamed guts of patients with inflammatory bowel disease. J. Allergy Clin. Immunol. 2018, 142, 1537–1547 e1538. [Google Scholar] [CrossRef] [Green Version]
- Foussat, A.; Cottrez, F.; Brun, V.; Fournier, N.; Breittmayer, J.P.; Groux, H. A comparative study between T regulatory type 1 and CD4+CD25+ T cells in the control of inflammation. J. Immunol. 2003, 171, 5018–5026. [Google Scholar] [CrossRef] [Green Version]
- Fedorak, R.N.; Gangl, A.; Elson, C.O.; Rutgeerts, P.; Schreiber, S.; Wild, G.; Hanauer, S.B.; Kilian, A.; Cohard, M.; LeBeaut, A.; et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 2000, 119, 1473–1482. [Google Scholar] [CrossRef]
- Schreiber, S.; Fedorak, R.N.; Nielsen, O.H.; Wild, G.; Williams, C.N.; Nikolaus, S.; Jacyna, M.; Lashner, B.A.; Gangl, A.; Rutgeerts, P.; et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease IL-10 Cooperative Study Group. Gastroenterology 2000, 119, 1461–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braat, H.; Rottiers, P.; Hommes, D.W.; Huyghebaert, N.; Remaut, E.; Remon, J.P.; van Deventer, S.J.; Neirynck, S.; Peppelenbosch, M.P.; Steidler, L. A phase I trial with transgenic bacteria expressing interleukin-10 in Crohn’s disease. Clin. Gastroenterol. Hepatol. 2006, 4, 754–759. [Google Scholar] [CrossRef]
- Veenbergen, S.; Li, P.; Raatgeep, H.C.; Lindenbergh-Kortleve, D.J.; Simons-Oosterhuis, Y.; Farrel, A.; Costes, L.M.M.; Joosse, M.E.; van Berkel, L.A.; de Ruiter, L.F.; et al. IL-10 signaling in dendritic cells controls IL-1beta-mediated IFNgamma secretion by human CD4(+) T cells: Relevance to inflammatory bowel disease. Mucosal Immunol. 2019, 12, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Desreumaux, P.; Foussat, A.; Allez, M.; Beaugerie, L.; Hebuterne, X.; Bouhnik, Y.; Nachury, M.; Brun, V.; Bastian, H.; Belmonte, N.; et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology 2012, 143, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Brun, V.; Bastian, H.; Neveu, V.; Foussat, A. Clinical grade production of IL-10 producing regulatory Tr1 lymphocytes for cell therapy of chronic inflammatory diseases. Int. Immunopharmacol. 2009, 9, 609–613. [Google Scholar] [CrossRef]
- Feagan, B.G.; Greenberg, G.R.; Wild, G.; Fedorak, R.N.; Pare, P.; McDonald, J.W.; Dube, R.; Cohen, A.; Steinhart, A.H.; Landau, S.; et al. Treatment of ulcerative colitis with a humanized antibody to the alpha4beta7 integrin. N. Engl. J. Med. 2005, 352, 2499–2507. [Google Scholar] [CrossRef]
- Panes, J.; Sandborn, W.J.; Schreiber, S.; Sands, B.E.; Vermeire, S.; D’Haens, G.; Panaccione, R.; Higgins, P.D.R.; Colombel, J.F.; Feagan, B.G.; et al. Tofacitinib for induction and maintenance therapy of Crohn’s disease: Results of two phase IIb randomised placebo-controlled trials. Gut 2017, 66, 1049–1059. [Google Scholar] [CrossRef]
- Danese, S.; Furfaro, F.; Vetrano, S. Targeting S1P in Inflammatory Bowel Disease: New Avenues for Modulating Intestinal Leukocyte Migration. J. Crohns Colitis 2018, 12, S678–S686. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 269–278. [Google Scholar] [CrossRef] [Green Version]
- Hyams, J.; Crandall, W.; Kugathasan, S.; Griffiths, A.; Olson, A.; Johanns, J.; Liu, G.; Travers, S.; Heuschkel, R.; Markowitz, J.; et al. Induction and maintenance infliximab therapy for the treatment of moderate-to-severe Crohn’s disease in children. Gastroenterology 2007, 132, 863–873; quiz 1165–1166. [Google Scholar] [CrossRef] [PubMed]
- Crandall, W.; Hyams, J.; Kugathasan, S.; Griffiths, A.; Zrubek, J.; Olson, A.; Liu, G.; Heuschkel, R.; Markowitz, J.; Cohen, S.; et al. Infliximab therapy in children with concurrent perianal Crohn disease: Observations from REACH. J. Pediatr. Gastroenterol. Nutr. 2009, 49, 183–190. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells 2020, 9, 110. https://doi.org/10.3390/cells9010110
Tindemans I, Joosse ME, Samsom JN. Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells. 2020; 9(1):110. https://doi.org/10.3390/cells9010110
Chicago/Turabian StyleTindemans, Irma, Maria E. Joosse, and Janneke N. Samsom. 2020. "Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD" Cells 9, no. 1: 110. https://doi.org/10.3390/cells9010110
APA StyleTindemans, I., Joosse, M. E., & Samsom, J. N. (2020). Dissecting the Heterogeneity in T-Cell Mediated Inflammation in IBD. Cells, 9(1), 110. https://doi.org/10.3390/cells9010110