The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Functions of CD38
2.1. Enzymatic Activity
2.2. Receptor/Ligand Activity
3. CD38 in Hyper-Inflammatory Responses
4. The Role of CD38 in Tumors
4.1. Hematological Malignancies
4.2. Solid Tumors
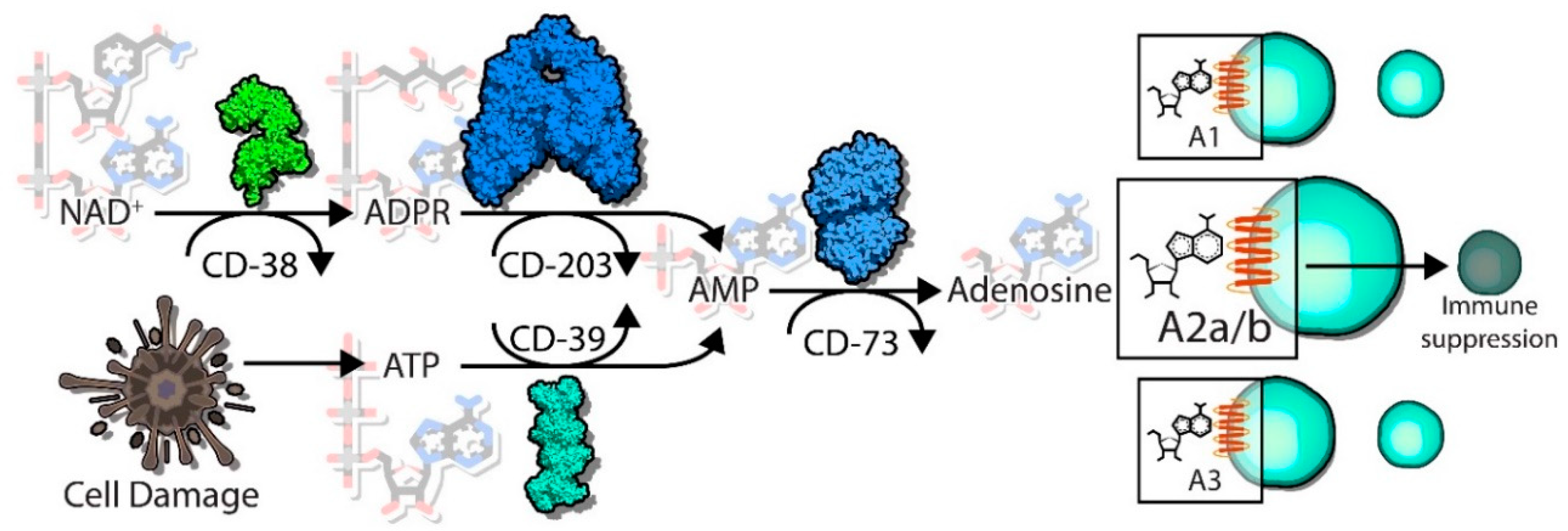
5. Adenosine Generating Pathways
5.1. CD39/CD73
5.2. CD38/CD203a/CD73
6. Immunosuppressive Functions of Adenosine-Producing Pathways
7. Targeting Adenosine-Generating Pathways
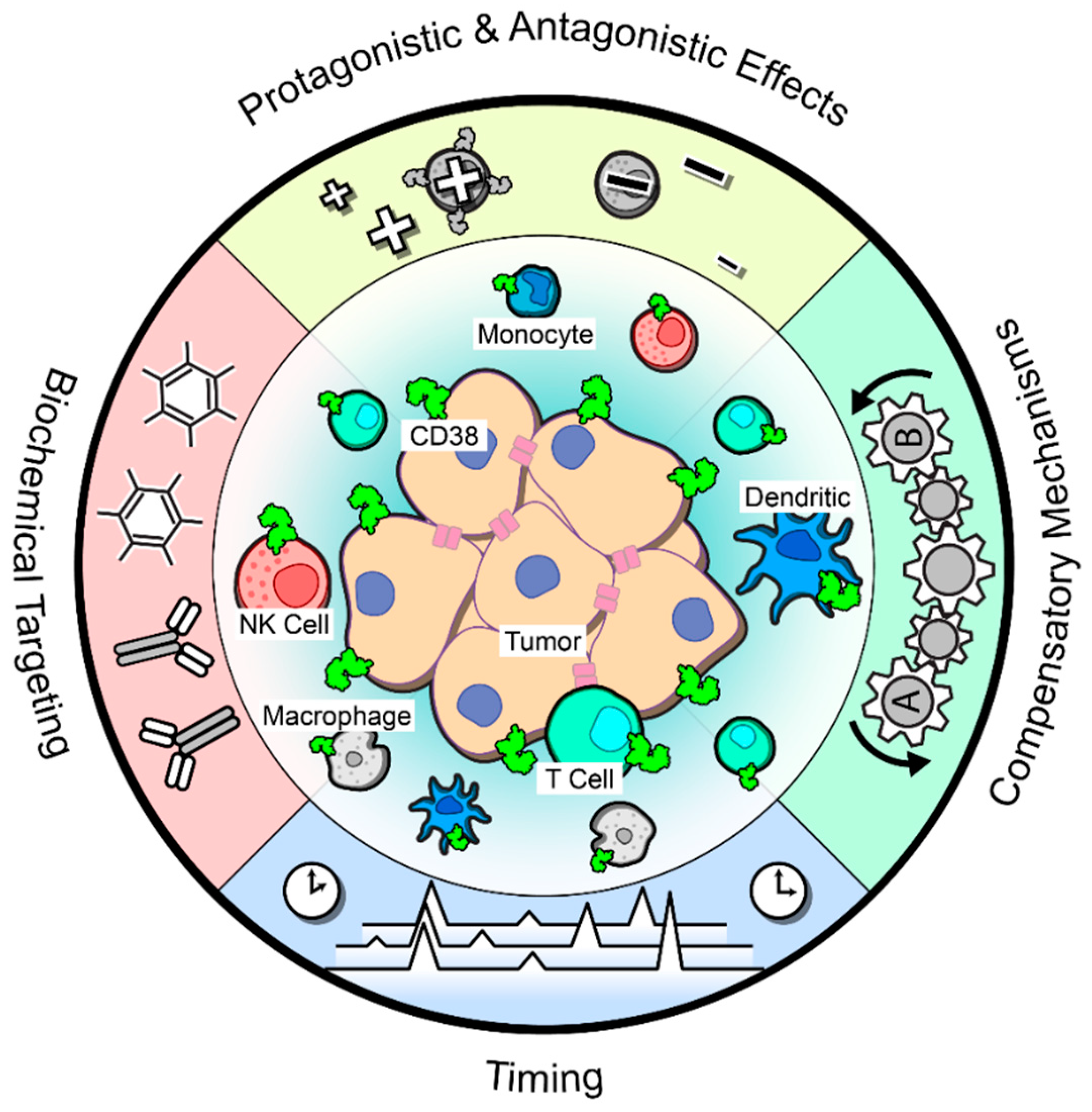
8. Remaining Questions Surrounding CD38 in Solid Tumors
8.1. Pro- and Antagonistic Effects
8.2. Biochemical Targeting
8.3. Compensatory Mechanisms
8.4. Effects of Timing
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kazemi, T.; Younesi, V.; Jadidi-Niaragh, F.; Yousefi, M. Immunotherapeutic approaches for cancer therapy: An updated review. Artif. Cells Nanomed. Biotechnol. 2016, 44, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: Mechanism, combinations, and clinical outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: Breakthroughs or backups. Nat. Immunol. 2019, 20, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Thallinger, C.; Füreder, T.; Preusser, M.; Heller, G.; Müllauer, L.; Höller, C.; Prosch, H.; Frank, N.; Swierzewski, R.; Berger, W.; et al. Review of cancer treatment with immune checkpoint inhibitors. Wien. Klin. Wochenschr. 2018, 130, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Emens, L.A. Targeting adenosine for cancer immunotherapy. J. Immunother. Cancer 2018, 6, 57. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [Green Version]
- Terhorst, C.; van Agthoven, A.; LeClair, K.; Snow, P.; Reinherz, E.; Schlossman, S. Biochemical studies of the human thymocyte cell-surface antigens T6, T9 and T10. Cell 1981, 23, 771–780. [Google Scholar] [CrossRef]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Levey, R.H.; Schlossman, S.F. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. USA 1980, 77, 1588–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavasi, F.; Funaro, A.; Roggero, S.; Horenstein, A.; Calosso, L.; Mehta, K. Human CD38: A glycoprotein in search of a function. Immunol. Today 1994, 15, 95–97. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Cockayne, D.A.; Muchamuel, T.; Grimaldi, J.C.; Muller-Steffner, H.L.N.; Randall, T.D.; Lund, F.E.; Murray, R.; Schuber, F.; Howard, M.C. Mice deficient for the Ecto-nicotinamide adenine dinucleotide glycohydrolase CD38 exhibit altered humoral immune responses. Blood 1998, 92, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Partida-Sánchez, S.; Goodrich, S.; Kusser, K.; Oppenheimer, N.; Randall, T.D.; Lund, F.E. Regulation of dendritic cell trafficking by the ADP-Ribosyl cyclase CD38: Impact on the development of humoral immunity. Immunity 2004, 20, 279–291. [Google Scholar] [CrossRef] [Green Version]
- Burgler, S. Role of CD38 expression in diagnosis and pathogenesis of chronic lymphocytic leukemia and its potential as therapeutic target. Crit. Rev. Immunol. 2015, 35, 417–432. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Damle, R.; Cutrona, G.; Ferrarini, M.; Chiorazzi, N. CD38 and chronic lymphocytic leukemia: A decade later. Blood 2011, 118, 3470–3478. [Google Scholar] [CrossRef] [Green Version]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A target for immunotherapeutic approaches in multiple myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef] [Green Version]
- Horenstein, A.L.; Bracci, C.; Morandi, F.; Malavasi, F. CD38 in adenosinergic pathways and metabolic re-programming in human multiple myeloma cells: In-tandem insights from basic science to therapy. Front. Immunol. 2019, 10, 760. [Google Scholar] [CrossRef]
- Du, Y.; Dai, Q.; Zhang, H.; Li, Q.; Song, K.; Fu, Y.; Min, W.; Liu, Z.; Li, R. CD38 deficiency downregulates the onset and pathogenesis of collagen-induced arthritis through the NF NF-κB Pathway. J. Immunol. Res. 2019, 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Postigo, J.; Iglesias, M.; Cerezo-Wallis, D.; Rosal-Vela, A.; García-Rodríguez, S.; Zubiaur, M.; Sancho, J.; Merino, R.; Merino, J. Mice deficient in CD38 develop an attenuated form of collagen type ii-induced arthritis. PLoS ONE 2012, 7, e33534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The pharmacology of CD38/NADase: An emerging target in cancer and diseases of aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.A.; Guedes, A.G.P.; Lund, F.E.; Subramanian, S.; Walseth, T.F.; Kannan, M.S. CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacol. Ther. 2017, 172, 116–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [Green Version]
- Levy, A.; Blacher, E.; Vaknine, H.; Lund, F.E.; Stein, R.; Mayo, L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro-Oncology 2012, 14, 1037–1049. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.B.; Lee, J.S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef] [Green Version]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [Green Version]
- Schmid, F.; Bruhn, S.; Weber, K.; Mittrücker, H.-W.; Guse, A.H. CD38: A NAADP degrading enzyme. FEBS Lett. 2011, 585, 3544–3548. [Google Scholar] [CrossRef] [Green Version]
- Alessio, M.; Roggero, S.; Funaro, A.; De Monte, L.B.; Peruzzi, L.; Geuna, M.; Malavasi, F. CD38 molecule: Structural and biochemical analysis on human T lymphocytes, thymocytes, and plasma cells. J. Immunol. 1990, 145, 878–884. [Google Scholar]
- Dianzani, U.; Funaro, A.; DiFranco, D.; Garbarino, G.; Bragardo, M.; Redoglia, V.; Buonfiglio, D.; De Monte, L.B.; Pileri, A.; Malavasi, F. Interaction between endothelium and CD4+CD45RA+ lymphocytes. Role of the human CD38 molecule. J. Immunol. 1994, 153, 952–959. [Google Scholar] [PubMed]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- Mehta, K.; Shahid, U.; Malavasi, F. Human CD38, a cell-surface protein with multiple functions. FASEB J. 1996, 10, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Zubiaur, M.; Izquierdo, M.; Terhorst, C.; Malavasi, F.; Sancho, J. CD38 ligation results in activation of the Raf-1/mitogen-activated protein kinase and the CD3-zeta/zeta-associated protein-70 signaling pathways in Jurkat T lymphocytes. J. Immunol. 1997, 159, 193–205. [Google Scholar]
- Lande, R.; Urbani, F.; Di Carlo, B.; Sconocchia, G.; Deaglio, S.; Funaro, A.; Malavasi, F.; Ausiello, C.M. CD38 ligation plays a direct role in the induction of IL-1β, IL-6, and IL-10 secretion in resting human monocytes. Cell. Immunol. 2002, 220, 30–38. [Google Scholar] [CrossRef]
- Ausiello, C.M.; Urbani, F.; la Sala, A.; Funaro, A.; Malavasi, F. CD38 ligation induces discrete cytokine mRNA expression in human cultured lymphocytes. Eur. J. Immunol. 1995, 25, 1477–1480. [Google Scholar] [CrossRef]
- Reinis, M.; Morra, M.; Funaro, A.; Di Primio, R.; Malavasi, F. Functional associations of CD38 with CD3 on the T-cell membrane. J. Biol. Regul. Homeost. Agents 1997, 11, 137–142. [Google Scholar]
- Munoz, P.; Navarro, M.D.; Pavon, E.J.; Salmeron, J.; Malavasi, F.; Sancho, J.; Zubiaur, M. CD38 signaling in T cells is initiated within a subset of membrane rafts containing Lck and the CD3-zeta subunit of the T cell antigen receptor. J. Biol. Chem. 2003, 278, 50791–50802. [Google Scholar] [CrossRef] [Green Version]
- Zubiaur, M.; Fernández, O.; Ferrero, E.; Salmerón, J.; Malissen, B.; Malavasi, F.; Sancho, J. CD38 is associated with lipid rafts and upon receptor stimulation leads to Akt/Protein Kinase B and Erk Activation in the absence of the CD3-ζ immune receptor tyrosine-based activation motifs. J. Biol. Chem. 2002, 277, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Funaro, A.; De Monte, L.B.; Dianzani, U.; Forni, M.; Malavasi, F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur. J. Immunol. 1993, 23, 2407–2411. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.-T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.H.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Zubiaur, M.; Gregorini, A.; Bottarel, F.; Ausiello, C.M.; Dianzani, U.; Sancho, J.; Malavasi, F. Human CD38 and CD16 are functionally dependent and physically associated in natural killer cells. Blood 2002, 99, 2490–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sconocchia, G.; Titus, J.A.; Mazzoni, A.; Visintin, A.; Pericle, F.; Hicks, S.W.; Malavasi, F.; Segal, D.M. CD38 triggers cytotoxic responses in activated human natural killer cells. Blood 1999, 94, 3864–3871. [Google Scholar] [CrossRef] [PubMed]
- Mallone, R.; Funaro, A.; Zubiaur, M.; Baj, G.; Ausiello, C.M.; Tacchetti, C.; Sancho, J.; Grossi, C.; Malavasi, F. Signaling through CD38 induces NK cell activation. Int. Immunol. 2001, 13, 397–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funaro, A.; Morra, M.; Calosso, L.; Zini, M.G.; Ausiello, C.M.; Malavasi, F. Role of the human CD38 molecule in B cell activation and proliferation. Tissue Antigens 1997, 49, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, T.; Hazeki, K.; Tsujimoto, N.; Inoue, S.-I.; Kurosu, H.; Kontani, K.; Hazeki, O.; Ui, M.; Katada, T. Association of phosphatidylinositol 3-kinase with the photo-oncogene product Cbl upon CD38 ligation by a specific monoclonal antibody in THP-1 cells. FEBS Lett. 1996, 397, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Kenji, K.; Kukimoto, I.; Nishina, H.; Hoshino, S.-I.; Hazeki, O.; Kanaho, Y.; Katada, T. Tyrosine phosphorylation of the c-cbl proto-oncogene product mediated by cell surface antigen CD38 in HL-60 cells. J. Biol. Chem. 1996, 271, 1534–1537. [Google Scholar] [CrossRef] [Green Version]
- Tsujimoto, N.; Kontain, K.; Inoue, S.-I.; Hoshino, S.-I.; Hazeki, O.; Malavasi, F.; Katada, T. Potentiation of chemotactic peptide-induced superoxide generation by CD38 ligation in human myeloid cell Lines1. J. Biochem. 1997, 121, 949–956. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Ferrero, E.; Funaro, A.; Sancho, J.; Ausiello, C.M.; Ortolan, E.; Vaisitti, T.; Zubiaur, M.; Fedele, G.; et al. CD38 and CD157 as receptors of the immune system: A bridge between innate and adaptive immunity. Mol. Med. 2006, 12, 334–341. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, D.A.; Dogan, S.; Walseth, T.F.; Miller, S.M.; Amrani, Y.; Panettieri, R.A.; Kannan, M.S. Modulation of calcium signaling by interleukin-13 in human airway smooth muscle: Role of CD38/cyclic adenosine diphosphate ribose pathway. Am. J. Respir. Cell Mol. Biol. 2004, 31, 36–42. [Google Scholar] [CrossRef]
- Guedes, A.G.; Jude, J.A.; Paulin, J.; Rivero-Nava, L.; Kita, H.; Lund, F.E.; Kannan, M.S. Airway responsiveness in CD38-deficient mice in allergic airway disease: Studies with bone marrow chimeras. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L485–L493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, A.G.P.; Paulin, J.; Rivero-Nava, L.; Kita, H.; Lund, F.E.; Kannan, M.S. CD38-deficient mice have reduced airway hyperresponsiveness following IL-13 challenge. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1286–L1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavoni, I.; Scagnolari, C.; Horenstein, A.L.; Leone, P.; Pierangeli, A.; Malavasi, F.; Ausiello, C.M.; Fedele, G. CD38 modulates respiratory syncytial virus-driven proinflammatory processes in human monocyte-derived dendritic cells. Immunology 2018, 154, 122–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhurst, J.A.; Lea, S.; Hardaker, E.; Dungwa, J.V.; Ravi, A.K.; Singh, D. Characterisation of lung macrophage subpopulations in COPD patients and controls. Sci. Rep. 2017, 7, 7143. [Google Scholar] [CrossRef] [Green Version]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [CrossRef]
- Matrai, Z. CD38 as a prognostic marker in CLL. Hematology 2005, 10, 39–46. [Google Scholar] [CrossRef]
- Durig, J.; Naschar, M.; Schmucker, U.; Renzing-Kohler, K.; Holter, T.; Huttmann, A.; Duhrsen, U. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia 2002, 16, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Vaisitti, T.; Aydin, S.; Rossi, D.; Cottino, F.; Bergui, L.; D’Arena, G.; Bonello, L.; Horenstein, A.L.; Brennan, P.; Pepper, C.; et al. CD38 increases CXCL12-mediated signals and homing of chronic lymphocytic leukemia cells. Leukemia 2010, 24, 958–969. [Google Scholar] [CrossRef]
- Pepper, C.; Lin, T.T.; Pratt, G.; Hewamana, S.; Brennan, P.; Hiller, L.; Hills, R.; Ward, R.; Starczynski, J.; Austen, B.; et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood 2008, 112, 3807–3817. [Google Scholar] [CrossRef]
- Damle, R.N.; Temburni, S.; Calissano, C.; Yancopoulos, S.; Banapour, T.; Sison, C.; Allen, S.L.; Rai, K.R.; Chiorazzi, N. CD38 expression labels an activated subset within chronic lymphocytic leukemia clones enriched in proliferating B cells. Blood 2007, 110, 3352–3359. [Google Scholar] [CrossRef] [Green Version]
- Ottaggio, L.; Viaggi, S.; Zunino, A.; Zupo, S.; Rossi, E.; Spriano, M.; Abbondandolo, A.; Ferrarini, M. Chromosome aberrations evaluated by comparative genomic hybridization in B-cell chronic lymphocytic leukemia: Correlation with CD38 expression. Haematologica 2003, 88, 769–777. [Google Scholar] [PubMed]
- Grubor, V.; Krasnitz, A.; Troge, J.E.; Meth, J.L.; Lakshmi, B.; Kendall, J.T.; Yamrom, B.; Alex, G.; Pai, D.; Navin, N.; et al. Novel genomic alterations and clonal evolution in chronic lymphocytic leukemia revealed by representational oligonucleotide microarray analysis (ROMA). Blood 2009, 113, 1294–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calissano, C.; Damle, R.N.; Hayes, G.; Murphy, E.J.; Hellerstein, M.K.; Moreno, C.; Sison, C.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; et al. In vivo intraclonal and interclonal kinetic heterogeneity in B-cell chronic lymphocytic leukemia. Blood 2009, 114, 4832–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, H.; Kawano, M.M.; Huang, N.; Harada, Y.; Iwato, K.; Tanabe, O.; Tanaka, H.; Sakai, A.; Asaoku, H.; Kuramoto, A. Phenotypic difference of normal plasma cells from mature myeloma cells. Blood 1993, 81, 2658–2663. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Owens, R.; Tricot, G.; Wilson, C.S. Flow cytometric immunophenotypic analysis of 306 cases of multiple myeloma. Am. J. Clin. Pathol. 2004, 121, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Overdijk, M.B.; Verploegen, S.; Bogels, M.; van Egmond, M.; Lammerts van Bueren, J.J.; Mutis, T.; Groen, R.W.; Breij, E.; Martens, A.C.; Bleeker, W.K.; et al. Antibody-mediated phagocytosis contributes to the anti-tumor activity of the therapeutic antibody daratumumab in lymphoma and multiple myeloma. mAbs 2015, 7, 311–321. [Google Scholar] [CrossRef]
- McKeage, K. Daratumumab: First global approval. Drugs 2016, 76, 275–281. [Google Scholar] [CrossRef]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune-regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [Green Version]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Van de Donk, N.W.C.J.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]
- U.S. Food & Drug Administration. Available online: https://www.fda.gov/drugs (accessed on 29 October 2019).
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK cells in daratumumab therapy for multiple myeloma overcome by Ex Vivo–Expanded autologous NK cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nijhof, I.S.; Groen, R.W.J.; Noort, W.A.; van Kessel, B.; de Jong-Korlaar, R.; Bakker, J.; van Bueren, J.J.L.; Parren, P.W.H.I.; Lokhorst, H.M.; van de Donk, N.W.C.J.; et al. Preclinical evidence for the therapeutic potential of CD38-targeted immuno-chemotherapy in multiple myeloma patients refractory to lenalidomide and bortezomib. Clin. Cancer Res. 2015, 21, 2802–2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahi, H.; Chrobok, M.; Gran, C.; Lund, J.; Gruber, A.; Gahrton, G.; Ljungman, P.; Wagner, A.K.; Alici, E. Infectious complications and NK cell depletion following daratumumab treatment of Multiple Myeloma. PLoS ONE 2019, 14, e0211927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karakasheva, T.A.; Dominguez, G.A.; Hashimoto, A.; Lin, E.W.; Chiu, C.; Sasser, K.; Lee, J.W.; Beatty, G.L.; Gabrilovich, D.I.; Rustgi, A.K. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight 2018, 3, e97022. [Google Scholar] [CrossRef]
- Blacher, E.; Ben Baruch, B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer 2015, 136, 1422–1433. [Google Scholar] [CrossRef]
- Bu, X.; Kato, J.; Hong, J.A.; Merino, M.J.; Schrump, D.S.; Lund, F.E.; Moss, J. CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 2018, 39, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Xiao, S.; Zhu, G.; Zheng, D.; He, J.; Pei, Z.; Li, G.; Zhou, Y. CD38 is highly expressed and affects the PI3K/Akt signaling pathway in cervical cancer. Oncol. Rep. 2014, 32, 2703–2709. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Xiao, S.; Chen, H.; Zhang, M.; Chen, Z.; Long, Y.; Gao, L.; Zhu, G.; He, J.; Peng, S.; et al. CD38 enhances the proliferation and inhibits the apoptosis of cervical cancer cells by affecting the mitochondria functions. Mol. Carcinog. 2017, 56, 2245–2257. [Google Scholar] [CrossRef]
- Liu, X.; Grogan, T.R.; Hieronymus, H.; Hashimoto, T.; Mottahedeh, J.; Cheng, D.; Zhang, L.; Huang, K.; Stoyanova, T.; Park, J.W.; et al. Low CD38 identifies progenitor-like inflammation-associated luminal cells that can initiate human prostate cancer and predict poor outcome. Cell Rep. 2016, 17, 2596–2606. [Google Scholar] [CrossRef]
- Mottahedeh, J.; Haffner, M.C.; Grogan, T.R.; Hashimoto, T.; Crowell, P.D.; Beltran, H.; Sboner, A.; Bareja, R.; Esopi, D.; Isaacs, W.B.; et al. CD38 is methylated in prostate cancer and regulates extracellular NAD+. Cancer Metab. 2018, 6, 13. [Google Scholar] [CrossRef]
- Chmielewski, J.P.; Bowlby, S.C.; Wheeler, F.B.; Shi, L.; Sui, G.; Davis, A.L.; Howard, T.D.; D’Agostino, R.B., Jr.; Miller, L.D.; Sirintrapun, S.J.; et al. CD38 Inhibits Prostate Cancer Metabolism and Proliferation by Reducing Cellular NAD(+) Pools. Mol. Cancer Res. MCR 2018, 16, 1687–1700. [Google Scholar] [CrossRef] [Green Version]
- Adriouch, S.; Haag, F.; Boyer, O.; Seman, M.; Koch-Nolte, F. Extracellular NAD(+): A danger signal hindering regulatory T cells. Microbes Infect. 2012, 14, 1284–1292. [Google Scholar] [CrossRef]
- Goding, J.W. Ecto-enzymes: Physiology meets pathology. J. Leukoc. Biol. 2000, 67, 285–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goding, J.W.; Terkeltaub, R.; Maurice, M.; Deterre, P.; Sali, A.; Belli, S.I. Ecto-phosphodiesterase/pyrophosphatase of lymphocytes and non-lymphoid cells: Structure and function of the PC-1 family. Immunol. Rev. 1998, 161, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, J.H.; Thompson, L.F.; Mueller, C.; Waickman, A.T.; Jalkanen, S.; Niemela, J.; Airas, L.; Bynoe, M.S. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2008, 105, 9325–9330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemelä, J.; Ifergan, I.; Yegutkin, G.G.; Jalkanen, S.; Prat, A.; Airas, L. IFN-β regulates CD73 and adenosine expression at the blood–brain barrier. Eur. J. Immunol. 2008, 38, 2718–2726. [Google Scholar] [CrossRef]
- Nikolova, M.; Carriere, M.; Jenabian, M.-A.; Limou, S.; Younas, M.; Kök, A.; Huë, S.; Seddiki, N.; Hulin, A.; Delaneau, O.; et al. CD39/Adenosine pathway is involved in AIDS progression. PLoS Pathog. 2011, 7, e1002110. [Google Scholar] [CrossRef] [Green Version]
- Hausler, S.F.; del Barrio, I.M.; Strohschein, J.; Chandran, P.A.; Engel, J.B.; Honig, A.; Ossadnik, M.; Horn, E.; Fischer, B.; Krockenberger, M.; et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine-generating enzymes responsible for adenosine receptor 2A-dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol. Immunother. CII 2011, 60, 1405–1418. [Google Scholar] [CrossRef]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015, 6, 25602–25618. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcotte, M.; Spring, K.; Pommey, S.; Chouinard, G.; Cousineau, I.; George, J.; Chen, G.M.; Gendoo, D.M.; Haibe-Kains, B.; Karn, T.; et al. CD73 is associated with poor prognosis in high-grade serous ovarian cancer. Cancer Res. 2015, 75, 4494–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.L.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Ngiow, S.F.; Madore, J.; Reinhardt, J.; Landsberg, J.; Chitsazan, A.; Rautela, J.; Bald, T.; Barkauskas, D.S.; Ahern, E.; et al. Targeting adenosine in BRAF-mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017, 77, 4684–4696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Yoshimura, K.; Kurabe, N.; Kahyo, T.; Kawase, A.; Tanahashi, M.; Ogawa, H.; Inui, N.; Funai, K.; Shinmura, K.; et al. Prognostic impact of CD73 and A2A adenosine receptor expression in non-small-cell lung cancer. Oncotarget 2017, 8, 8738–8751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.C.; Rhee, K.; Kim, W.B.; Cho, A.; Song, J.; Anker, J.F.; Oh, M.; Bais, P.; Namburi, S.; Chuang, J.; et al. Immunologic and clinical implications of CD73 expression in non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 12050. [Google Scholar] [CrossRef]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [Green Version]
- Perrot, I.; Michaud, H.-A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [Green Version]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Ngiow Shin, F.; Barkauskas Deborah, S.; Sult, E.; Hay, C.; Blake Stephen, J.; Huang, Q.; Liu, J.; Takeda, K.; Teng Michele, W.L.; et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell 2016, 30, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, N.; Bazhenova, L. New strategies in immunotherapy for lung cancer: Beyond PD-1/PD-L1. Ther. Adv. Respir. Dis. 2018, 12, 1753466618794133. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. Available online: http://clinicaltrials.gov (accessed on 13 September 19).
- Adriouch, S.; Hubert, S.; Pechberty, S.; Koch-Nolte, F.; Haag, F.; Seman, M. NAD+ released during inflammation participates in T cell homeostasis by inducing ART2-mediated death of naive T cells In Vivo. J. Immunol. 2007, 179, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J. Biol. Chem. 2006, 281, 31419–31429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Corriden, R.; Inoue, Y.; Yip, L.; Hashiguchi, N.; Zinkernagel, A.; Nizet, V.; Insel, P.A.; Junger, W.G. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 2006, 314, 1792–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yao, Y.; Sumi, Y.; Li, A.; To, U.K.; Elkhal, A.; Inoue, Y.; Woehrle, T.; Zhang, Q.; Hauser, C.; et al. Purinergic signaling: A fundamental mechanism in neutrophil activation. Sci. Signal. 2010, 3, ra45. [Google Scholar] [CrossRef] [Green Version]
- Adriouch, S.; Ohlrogge, W.; Haag, F.; Koch-Nolte, F.; Seman, M. Rapid induction of naive T cell apoptosis by Ecto-nicotinamide adenine dinucleotide: Requirement for Mono(ADP-Ribosyl)Transferase 2 and a downstream effector. J. Immunol. 2001, 167, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Seman, M.; Adriouch, S.; Scheuplein, F.; Krebs, C.; Freese, D.; Glowacki, G.; Deterre, P.; Haag, F.; Koch-Nolte, F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 2003, 19, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Hubert, S.; Rissiek, B.; Klages, K.; Huehn, J.; Sparwasser, T.; Haag, F.; Koch-Nolte, F.; Boyer, O.; Seman, M.; Adriouch, S. Extracellular NAD+ shapes the Foxp3+ regulatory T cell compartment through the ART2-P2X7 pathway. J. Exp. Med. 2010, 207, 2561–2568. [Google Scholar] [CrossRef]
- The ASCO Post. Available online: http://www.ascopost.com/news/58952 (accessed on 5 October 19).
- Casneuf, T.; Xu, X.S.; Adams, H.C., 3rd; Axel, A.E.; Chiu, C.; Khan, I.; Ahmadi, T.; Yan, X.; Lonial, S.; Plesner, T.; et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017, 1, 2105–2114. [Google Scholar] [CrossRef] [PubMed]
- Frasca, L.; Fedele, G.; Deaglio, S.; Capuano, C.; Palazzo, R.; Vaisitti, T.; Malavasi, F.; Ausiello, C.M. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 2006, 107, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhu, W.; Chen, Y.; Lin, Z.; Ma, S. CD38 gene-modified dendritic cells inhibit murine asthma development by increasing IL-12 production and promoting Th1 cell differentiation. Mol. Med. Rep. 2016, 14, 4374–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Wetzel, M.C.; Bartle, L.M.; Skaletskaya, A.; Goldmacher, V.S.; Vallee, F.; Zhou-Liu, Q.; Ferrari, P.; Pouzieux, S.; Lahoute, C.; et al. SAR650984, a novel humanized CD38-targeting antibody, demonstrates potent antitumor activity in models of multiple myeloma and other CD38+ hematologic malignancies. Clin. Cancer Res. 2014, 20, 4574–4583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, C.D.; Becherer, J.D.; Boros, E.E.; Cadilla, R.; Carpenter, T.; Cowan, D.; Deaton, D.N.; Guo, Y.; Harrington, W.; Henke, B.R.; et al. Discovery, synthesis, and biological evaluation of Thiazoloquin(az)olin(on)es as potent CD38 inhibitors. J. Med. Chem. 2015, 58, 3548–3571. [Google Scholar] [CrossRef]
- Becherer, J.D.; Boros, E.E.; Carpenter, T.Y.; Cowan, D.J.; Deaton, D.N.; Haffner, C.D.; Jeune, M.R.; Kaldor, I.W.; Poole, J.C.; Preugschat, F.; et al. Discovery of 4-Amino-8-quinoline carboxamides as novel, submicromolar inhibitors of NAD-hydrolyzing enzyme CD38. J. Med. Chem. 2015, 58, 7021–7056. [Google Scholar] [CrossRef]
- Pogue, S.L.; Taura, T.; Bi, M.; Yun, Y.; Sho, A.; Mikesell, G.; Behrens, C.; Sokolovsky, M.; Hallak, H.; Rosenstock, M.; et al. Targeting attenuated interferon-α to myeloma cells with a CD38 antibody induces potent tumor regression with reduced off-target activity. PLoS ONE 2016, 11, e0162472. [Google Scholar] [CrossRef]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; Lammerts van Bueren, J.J.; Parren, P.W.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.; Groen, R.W.J.; et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konen, J.M.; Fradette, J.J.; Gibbons, D.L. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells 2020, 9, 52. https://doi.org/10.3390/cells9010052
Konen JM, Fradette JJ, Gibbons DL. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells. 2020; 9(1):52. https://doi.org/10.3390/cells9010052
Chicago/Turabian StyleKonen, Jessica M., Jared J. Fradette, and Don L. Gibbons. 2020. "The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors" Cells 9, no. 1: 52. https://doi.org/10.3390/cells9010052
APA StyleKonen, J. M., Fradette, J. J., & Gibbons, D. L. (2020). The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells, 9(1), 52. https://doi.org/10.3390/cells9010052