Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence


 ,
,
Abstract
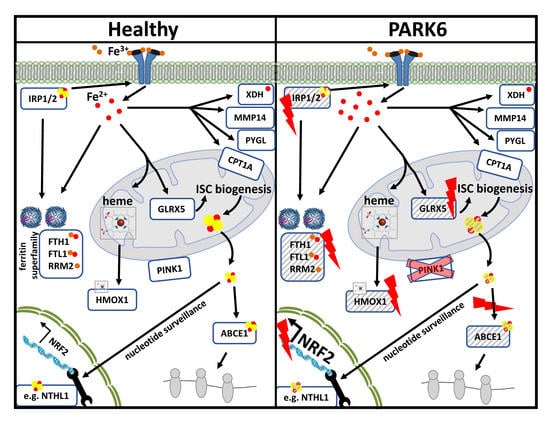
:
1. Introduction
2. Results
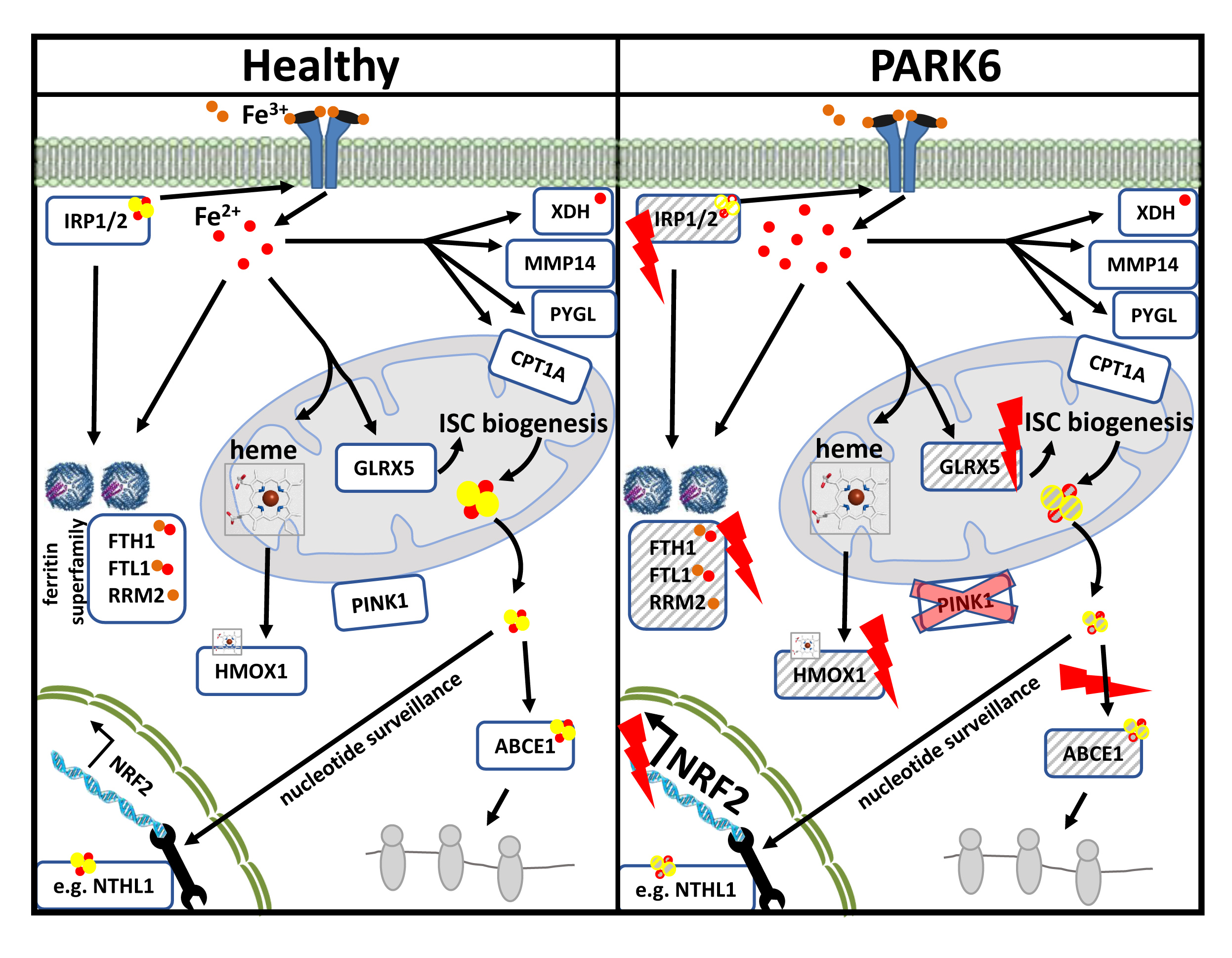
2.1. Global Proteome Profile Adaptations upon Iron Overload and Iron Depletion in WT and Pink1−/− Mouse Embryonic Fibroblast (MEF) Cells
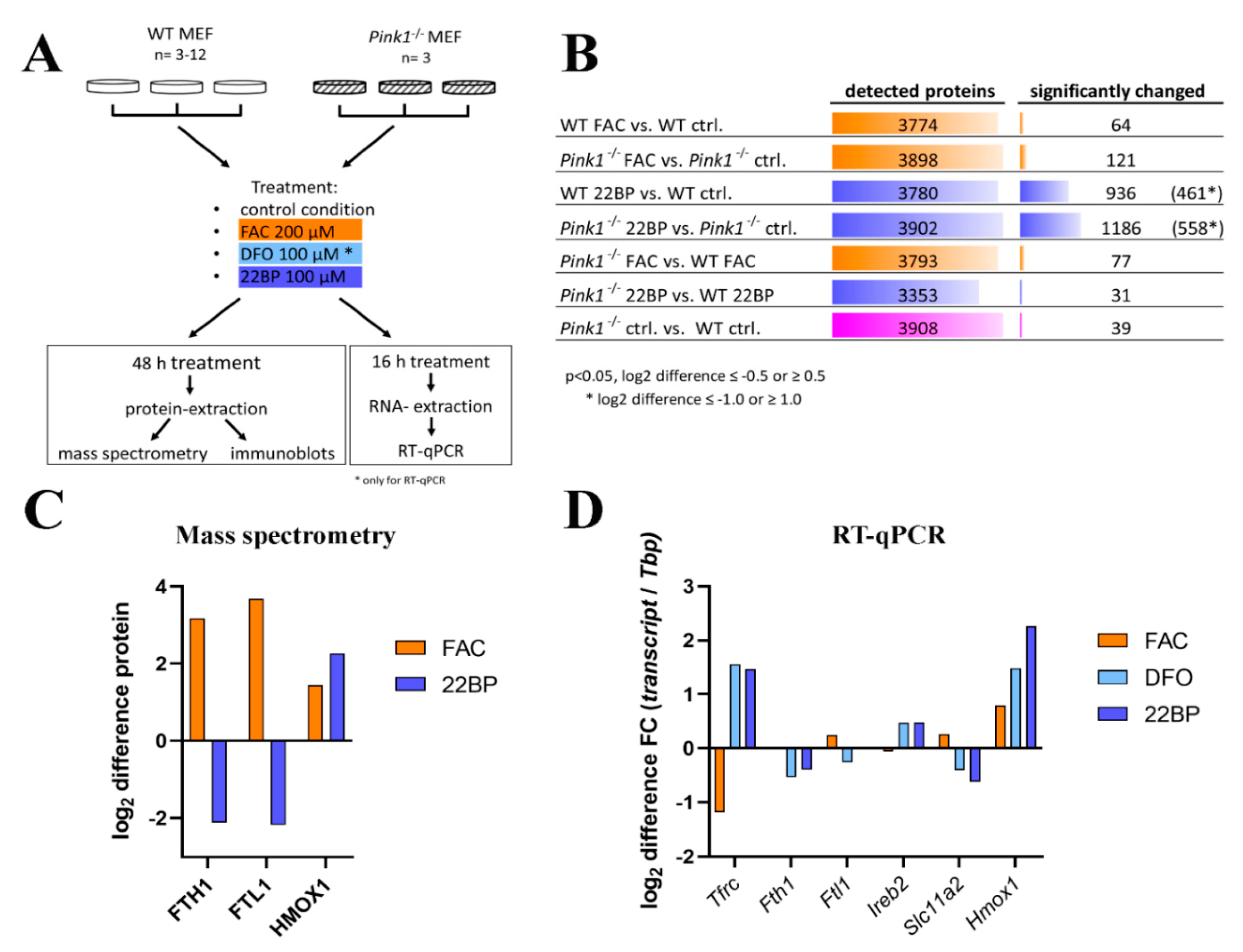
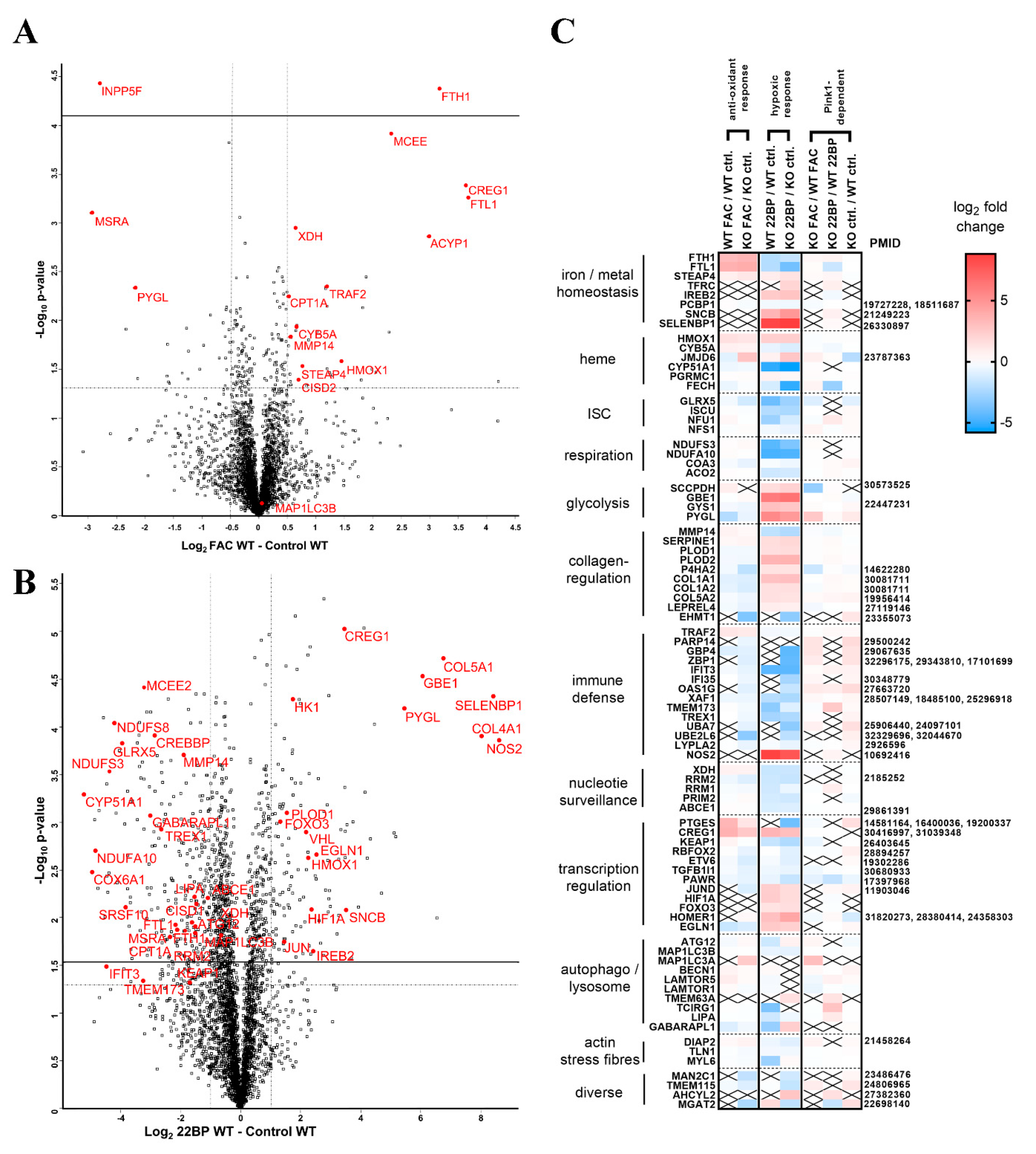
2.1.1. FAC-Effects
2.1.2. 22BP-Effects
2.1.3. Pink1−/− Effects
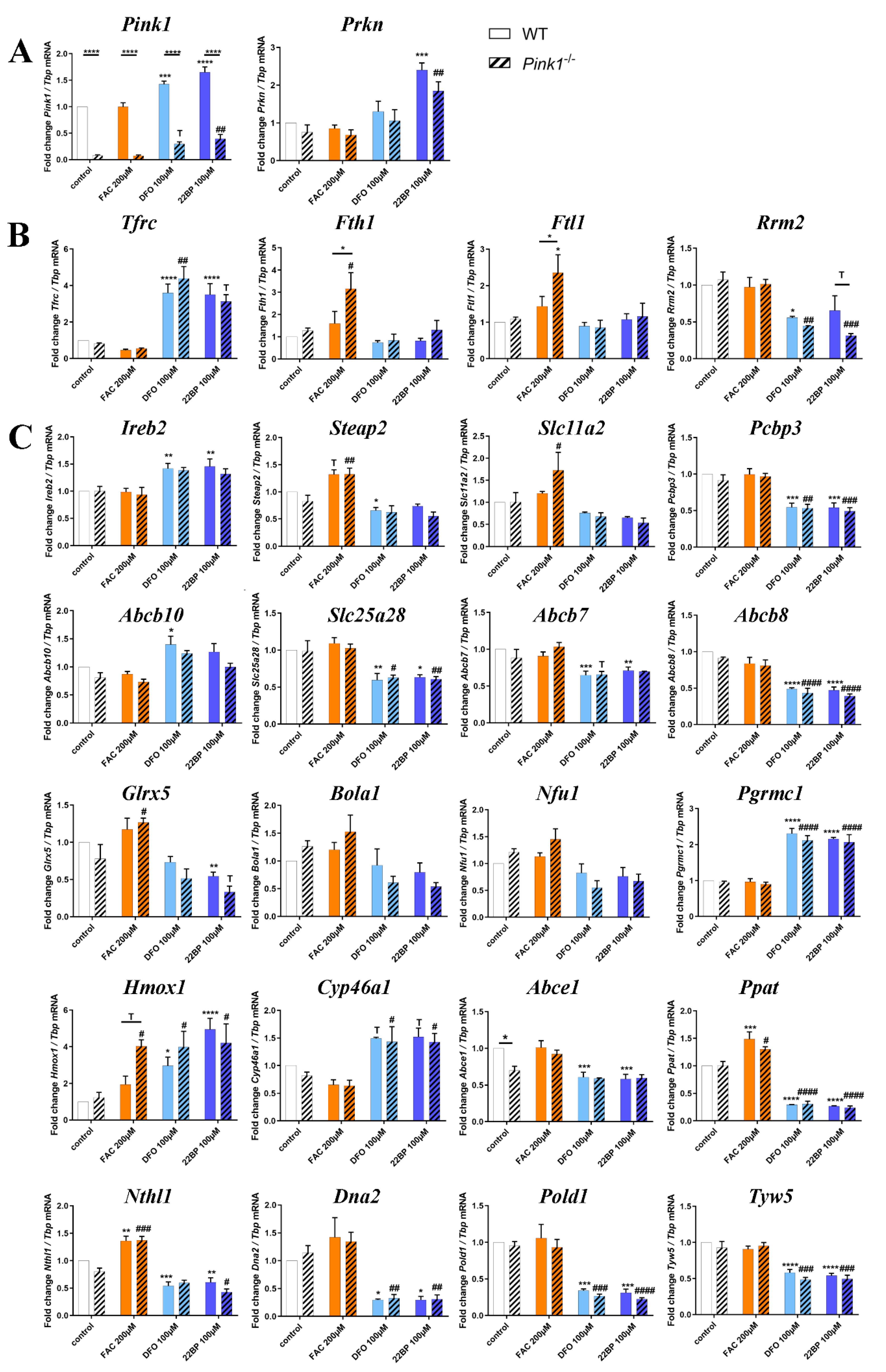
2.2. Transcriptional Analyses of Cellular Iron Homeostasis Factors in WT MEF in Response to Altered Iron Levels
2.3. Transcriptional Analyses of Expression Adaptations of Pink1−/− MEF to Altered Iron Levels
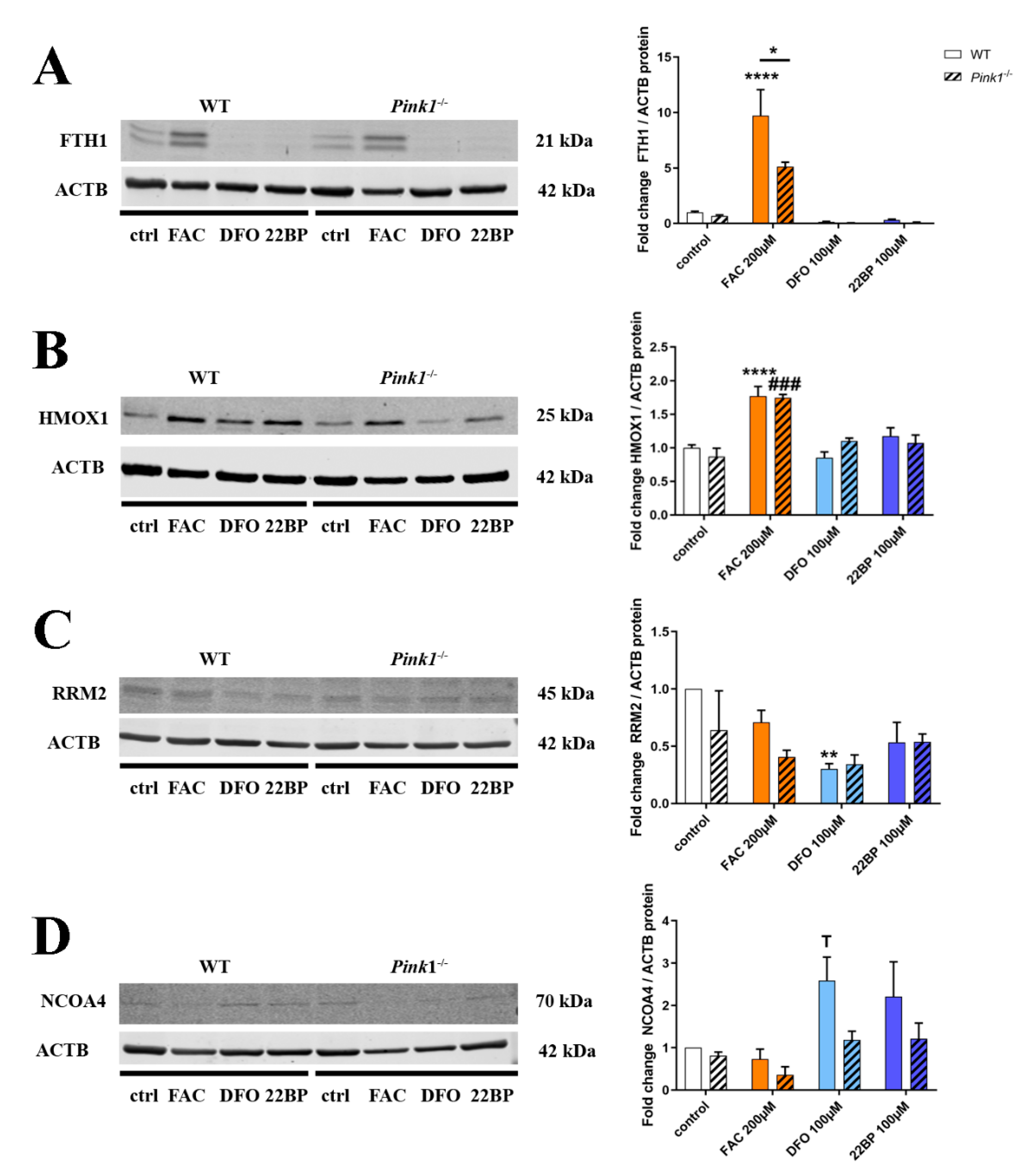
2.4. Quantitative Immunoblots for Validation and Mechanistic Analyses
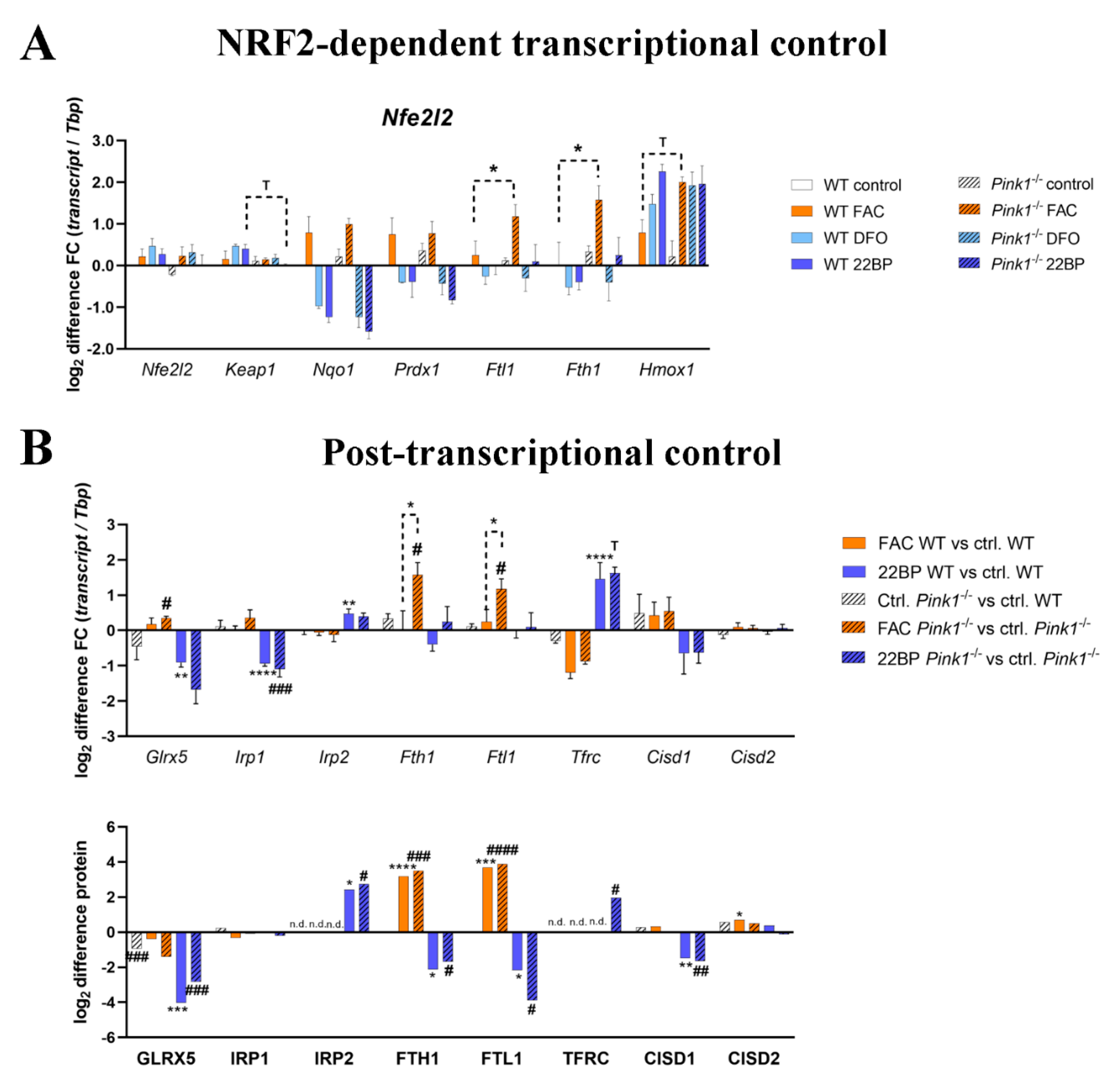
2.5. Regulation of Relevant Transcription Factors with Their Targets Is Subtly Modified by PINK1
2.6. Regulation of the Post-Transcriptional Control over Iron Homeostasis Is Modified by PINK1
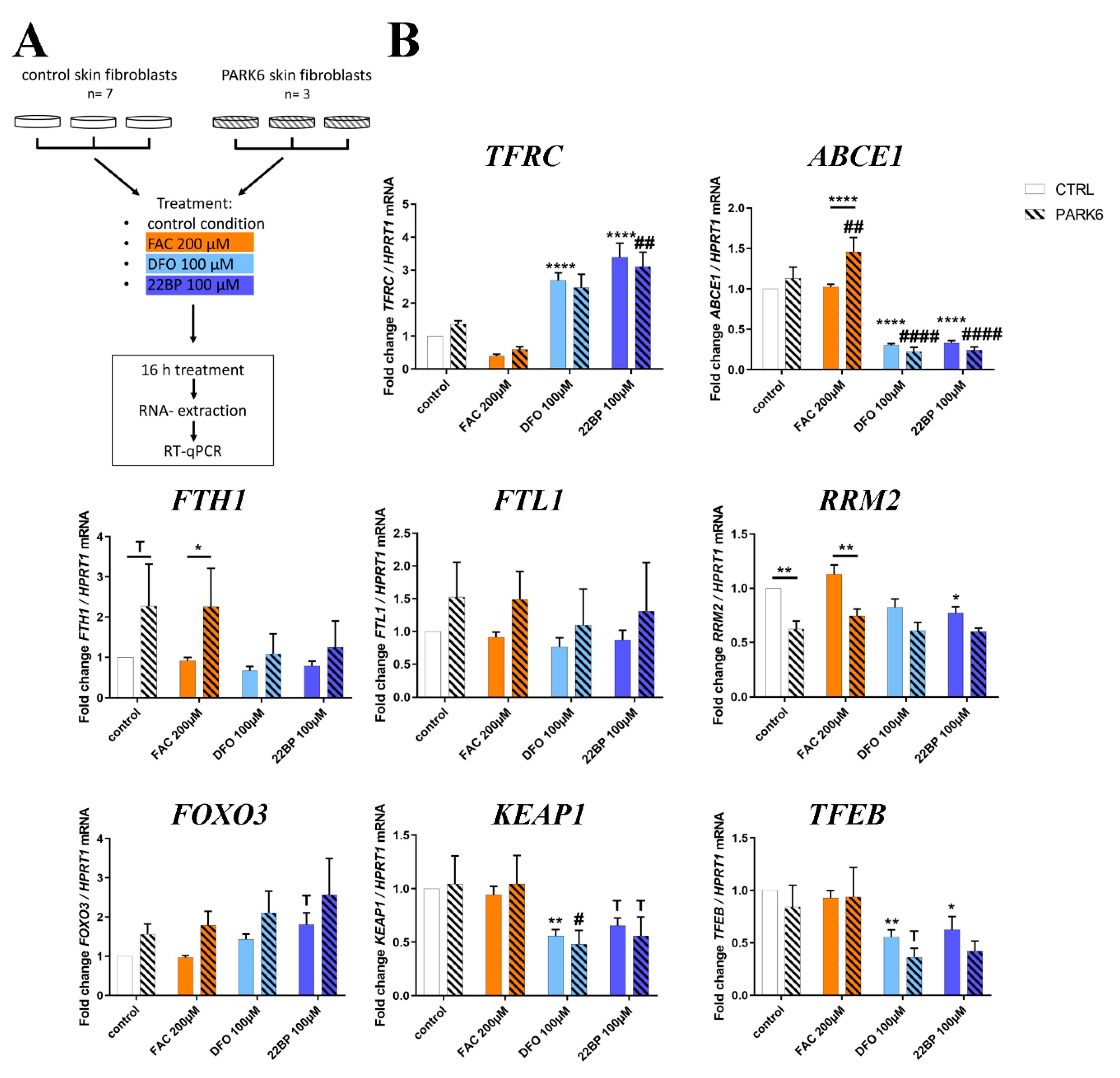
2.7. PINK1-Dependent Effects on Expression in Human Skin Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Mouse Embryonic Fibroblast Generation and Culture
4.2. Human Fibroblasts
4.3. Iron Overload/Depletion Experiments
4.4. Reverse Transcriptase Real-Time Quantitative PCR
4.5. Quantitative Immunoblotting
4.6. Proteomics Sample Preparation with Label-Free Quantification (LFQ)
4.7. LC-MS/MS Instrument Settings for Shotgun Proteome Profiling and Data Analysis
4.8. Statistical Evaluation
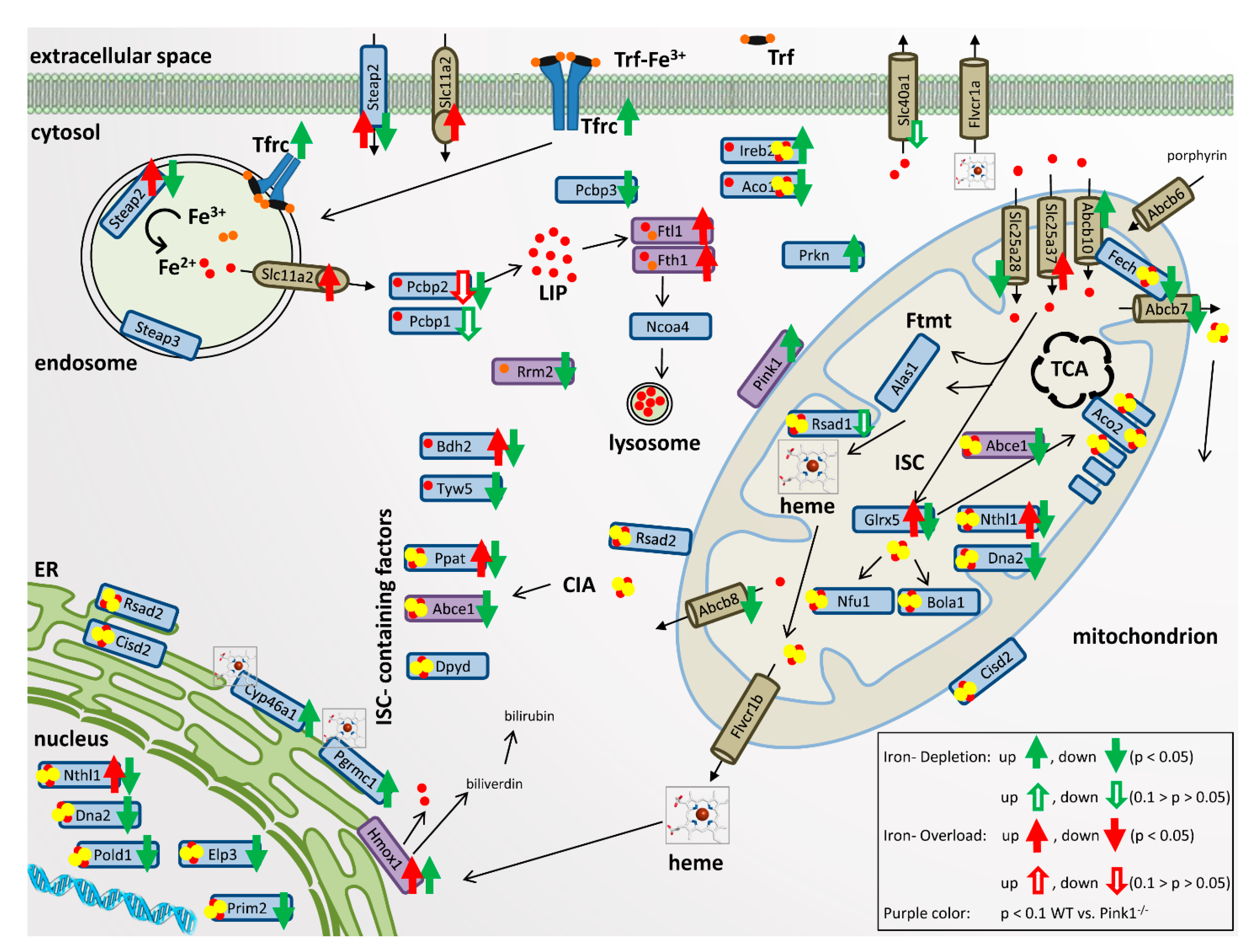
4.9. Visualization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| µM | MicroMolar |
| 22BP | 2,2′-Bipyridine |
| Abcb1 | ATP binding cassette subfamily B member 1 |
| Abcb7 | ATP binding cassette subfamily B member 7 |
| Abcb8 | ATP binding cassette subfamily B member 8 |
| Abcb10 | ATP binding cassette subfamily B member 10 |
| Abce1 | ATP binding cassette subfamily E member 1, RNase L inhibitor 1 |
| Aco1 | Aconitase 1, cytoplasmic, aka Irp1, aka Ferritin repressor protein |
| Aco2 | Aconitase 2, mitochondrial |
| ACSL3 | Acyl-CoA synthetase long-chain family member 3 |
| ACTB | Actin beta |
| ACYP1 | Acylphosphatase 1 |
| AGPAT1 | 1-Acylglycerol-3-Phosphate O-Acyltransferase 1 |
| AHCYL2 | Adenosylhomocysteinase like 2 |
| aka | also known as |
| ALA | 5′-aminolevulinic acid |
| Alas1 | 5′-aminolevulinate synthase 1 |
| AMDHD2 | Amidohydrolase domain containing 2 |
| ANOVA | Analysis of variance |
| ARMC1 | Armadillo repeat containing 1 |
| ARPP19 | CAMP regulated phosphoprotein 19 |
| ARNT | Aryl hydrocarbon receptor nuclear translocator |
| ATG4B | Autophagy related 4B cysteine peptidase |
| ATG12 | Autophagy related 12 |
| ATPase | Adenosintriphosphatase |
| Bdh2 | 3-hydroxybutyrate dehydrogenase-2 |
| BECN1 | Beclin 1, Autophagy related |
| Bola1 | BolA Family Member 1 |
| Brip1 | Brca1 Interacting Protein C-Terminal Helicase 1, Fancj |
| BUD31 | BUD31 homolog, Functional spliceosome-associated protein 17 |
| cAMP | Cyclic adenosine monophosphate |
| CCDC58 | Coiled-coil domain containing 58 |
| Cdc42bpa | Cdc42 binding protein kinase alpha (Dmpk-like), Mrcka |
| CIA | Cytosolic iron-sulfur cluster assembly machinery |
| Cisd1 | CDGSH iron-sulfur domain 1 |
| Cisd2 | CDGSH iron-sulfur domain 2, Wfs2 |
| CO2 | Carbon dioxide |
| COA3 | Cytochrome C oxidase assembly factor 3 |
| COL1A1/2 | Collagen type I alpha 1 chain, Collagen type I alpha 2 chain |
| COL4A1 | Collagen type IV alpha 1 chain |
| COL5A1 | Collagen type IV alpha 1 chain |
| COX1 | Mitochondrially encoded cytochrome C oxidase I |
| COX6A1 | Cytochrome C oxidase subunit 6A1, mitochondrial |
| Cp | Ceruloplasmin |
| CPT1A | Carnitine palmitoyltransferase 1A |
| CREBBP | CREB binding protein |
| CREG1 | Cellular repressor of E1A stimulated genes 1 |
| CYB5A | Cytochrome B5 type A |
| Cyp46a1 | Cytochrome P450 Family 46 Subfamily A Member 1 |
| Cyp56a1 | Cytochrome P450 Family 51 Subfamily A Member 1 |
| DFO | Deferoxamine |
| DIP2A | Disco interacting protein 2 homolog A |
| Dmt1 | Divalent metal transporter 1, encoded by Slc11a2 |
| DNA | Desoxyribonucleic acid |
| Dna2 | DNA replication helicase/nuclease 2 |
| Dpyd | Dihydropyrimidine dehydrogenase |
| Elp3 | Elongator acetyltransferase complex subunit 3 |
| EGLN1 | Egl-9 family hypoxia-inducible factor 1 |
| ER | Endoplasmic reticulum |
| Ercc2 | ERCC excision repair 2, TFIIH core complex helicase subunit, Cxpd |
| ETV6 | ETS variant transcription factor 6 |
| EXOSC4/10 | Exosome component 4/10 |
| FAC | Ferric ammonium acid |
| Fdx1 | Ferredoxin 1, mitochondrial Adrenodoxin |
| Fech | Ferrochelatase, Heme synthase |
| Flvcr1a | Feline leukemia virus subgroup C cellular receptor 1a, Pcarp |
| Flvcr1b | Feline leukemia virus subgroup C cellular receptor 1b, Pcarp |
| FOXO3 | Forkhead box O3 |
| Fth1 | Ferritin heavy chain |
| Ftl1 | Ferritin light chain |
| Ftmt | Mitochondrial ferritin |
| Fxn | Frataxin, aka Frda |
| g | Gram |
| GABARAPL1 | GABA type A receptor associated protein like 1 |
| GBE1 | Glucan (1,4-Alpha-), branching enzyme 1 |
| GBP4 | Guanylate binding protein 4 |
| Glrx5 | Glutaredoxin 5 |
| GPR126 | G-Protein coupled receptor 126 |
| GPSM2 | G Protein signaling modulator 2 |
| GYPC | Glycophorin C |
| GYS1 | Glycogen synthase 1 |
| h | Hour |
| Hbs1l | Hbs1 like translational GTPase, Erfs |
| Hebp1 | Heme binding protein 1 |
| HIF1A | Hypoxia-Inducible Factor 1-alpha |
| HK1 | Hexokinase 1 |
| HMGCL | Mitochondrial 3-Hydroxy-3-Methylglutaryl-CoA Lyase |
| Hmox1 | Heme oxygenase 1, HO-1 |
| HOMER1 | Homer scaffold protein 1 |
| IFIT3 | Interferon induced protein with tetratricopeptide repeats 3 |
| IKBKAP | IkappaB kinase complex-associated protein, Elp1 |
| IMPC | International mouse phenotyping consortium |
| INPP5F | Inositol Polyphosphate-5-Phosphatase F |
| IRE | Iron response element |
| Ireb1 | Iron-responsive element-binding protein 1 (Irp1), encoded by Aco1 |
| Ireb2 | Iron-responsive element-binding protein 2 (Irp2), encoded by Ireb2 |
| IRF2BP1 | Interferon regulatory factor 2 binding protein 1 |
| Irp1 | Iron regulatory protein 1 (cytosolic aconitase = Ireb1), encoded by Aco1 |
| Irp2 | Iron regulatory protein 2 (Aco3 = Ireb2), encoded by Ireb2 mRNA |
| ISC | Iron-sulfur-cluster |
| JMJD6 | Jumonji domain protein 6, Arg-demethylase, Lys-hydroxylase |
| JUN | Jun proto-oncogene, AP-1 transcription factor subunit |
| KEAP1 | Kelch-like ECH associated protein 1 |
| L | Liter |
| LAMTOR1/5 | Late endosomal/lysosomal adaptor, MAPK and MTOR activator 1/5 |
| LBH | LBH regulator of WNT signaling pathway |
| LFQ | Label free quantification |
| LIP | Labile iron pool |
| LIPA | Lipase A, lysosomal acid type |
| MAN2C1 | Mannosidase alpha class 2C member 1 |
| MAP1LC3A/B | Microtubule associated protein 1 light chain 3 alpha/beta |
| MCEE | Methylmalonyl-CoA Epimerase, mitochondrial |
| MEF | Mouse embryonic fibroblast |
| MEF2D | Myocyte enhancer factor 2D |
| Mfrn1 | Mitoferrin 1, encoded by Slc25a37 |
| Mfrn2 | Mitoferrin 2, encoded by Slc25a28 |
| Min | Minute |
| MMP14 | Matrix metallopeptidase 14 |
| MRPS36 | Mitochondrial ribosomal protein S36 |
| MSRA | Methionine sulfoxide reductase A |
| mRNA | Messenger ribonucleic acid |
| mTOR | Mechanistic target of rapamycin kinase |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| MYD88 | Myeloid differentiation primary response protein MyD88 |
| MYL6 | Myosin light chain 6 |
| Ncoa4 | Nuclear receptor coactivator 4 |
| NDUFA10 | NADH:Ubiquinone oxidoreductase subunit A10 |
| NDUFS3/8 | NADH:Ubiquinone oxidoreductase core subunit S3/subunit S8 |
| NFkB | Nuclear Factor ‘kappa-light-chain-enhancer’ of activated B-cells |
| Nfu1 | Nfu1 iron-sulfur cluster scaffold |
| NMRAL1 | NmrA like redox sensor 1 |
| NOS2 | Nitric oxide synthase 2 |
| Not4 | Ccr4-Not transcription complex subunit 4 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| NRF2 | Nuclear factor, erythroid 2 like 2, Nfe2l2 |
| NSMCE2 | Non-structural maintenance of chromosomes element 2 homolog |
| Nthl1 | Nth like DNA glycosylase 1 |
| NUCKS1 | Nuclear casein kinase and cyclin-dependent kinase substrate 1 |
| NUP160 | Nucleoporin 160 |
| OAS1G | 2′-5′-Oligoadenylate synthetase 1 |
| P4HA2 | Prolyl 4-hydroxylase subunit alpha 2 |
| PANK4 | Pantothenate kinase 4 |
| Parkin | Parkinson disease protein 2 |
| PAWR | Pro-apoptotic WT1 regulator |
| Pcbp1 | Poly(RC) binding protein 1, hnRNP-E1 |
| Pcbp2 | Poly(RC) binding protein 2, hnRNP-E2 |
| PD | Parkinson’s disease |
| PDDC1 | Glutamine amidotransferase like Class 1 domain containing 1, Gatd1 |
| Pgrmc1 | Progesterone receptor membrane component 1, Dap1 |
| Pink1 | PTEN induced kinase 1 |
| PLOD1 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 1 |
| PMPCA | Peptidase, mitochondrial processing subunit alpha |
| Pold1 | DNA polymerase delta 1 catalytic subunit, Cdc2 homolog |
| POMP | Proteasome maturation protein |
| Ppat | Phosphoribosyl pyrophosphate amidotransferase, Gpat, ATase |
| Prim2 | DNA primase subunit 2 |
| PTGES | Prostaglandin E synthase |
| PMID | PubMed database of medical literature, reference IDentifier number |
| PYGL | Glycogen Phosphorylase L |
| RBFOX2 | RNA binding fox-1 homolog 2 |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| RPF2 | Ribosome production factor 2 homolog |
| RRM1/2 | Ribonucleotide reductase regulatory subunit M1/M2 |
| rRNA | Ribosomal RNA |
| Rsad1 | Radical S-adenosyl methionine domain containing 1 |
| Rsad2 | Radical S-adenosyl methionine domain containing 2 |
| RT | Room temperature |
| RT-qPCR | reverse-transcriptase real-time quantitative polymerase chain reaction |
| Rtel1 | Regulator of telomere elongation helicase 1 |
| SCCPDH | Saccharopine dehydrogenase |
| SEM | Standard error of the mean |
| SELENBP1 | Selenium binding protein 1 |
| SERPINE2 | Serpin family E member 2 |
| SH2B1 | SH2 domain-containing protein 1B |
| Slc11a2 | Solute carrier family 11 member 2, Divalent metal transporter 1, Dmt1 |
| Slc25a37 | Solute carrier family 25 member 37, Mitoferrin 1 |
| Slc25a28 | Solute carrier family 25 member 28, Mitoferrin 2 |
| Slc40a1 | Solute carrier family 40 member 1, Ferroportin 1, Ireg1 |
| SNCB | Synuclein beta |
| SMX30 | Sorting nexin family member 30, ATG24A |
| SPP1 | Secreted phosphoprotein 1 |
| SRSF10 | Serine and arginine-rich splicing factor 10 |
| SSBP1 | Single-stranded DNA binding protein 1 |
| Steap2 | Six transmembrane epithelial antigen of the prostate 2, Stmp |
| Steap3 | Six transmembrane epithelial antigen of the prostate 3, Stmp3 |
| Steap4 | Six transmembrane epithelial antigen of the prostate 4, Stamp2 |
| STRING | Search Tool for the Retrieval of Interacting Genes/Proteins |
| SYN1 | Synapsin I |
| Tbp | Tata-binding protein |
| TBST | Tris-buffered saline/Tween 20 |
| TCA | Tricarboxylic acid cycle |
| TCIRG1 | T Cell immune regulator 1, ATPase H+ transporting V0 subunit A3 |
| TIMM10 | Translocase of inner mitochondrial membrane 10 |
| TMEM115 | Transmembrane protein 115 |
| TMEM173 | Stimulator of interferon genes protein, Sting |
| TMEM63A | Transmembrane protein 63A |
| TRAF2 | TNF receptor-associated factor 2 |
| TREX1 | Three prime repair exonuclease 1 |
| Trf | Transferrin |
| Tfrc | Transferrin receptor 1 |
| Tyw5 | tRNA wybutosine synthesizing protein 5 |
| UPP1 | Uridine phosphorylase 1 |
| UTR | Untranslated region |
| V | Volt |
| VHL | Von Hippel-Lindau tumor suppressor |
| WT | Wildtype |
| XAF1 | XIAP associated factor 1 |
| XDH | Xanthine dehydrogenase |
| ZBP1 | Z-DNA binding protein 1 |
References
- Weiland, A.; Wang, Y.; Wu, W.; Lan, X.; Han, X.; Li, Q.; Wang, J. Ferroptosis and Its Role in Diverse Brain Diseases. Mol. Neurobiol. 2019, 56, 4880–4893. [Google Scholar] [CrossRef] [PubMed]
- Stehling, O.; Lill, R. The Role of Mitochondria in Cellular Iron-Sulfur Protein Biogenesis: Mechanisms, Connected Processes, and Diseases. Cold Spring Harb. Perspect. Biol. 2013, 5, a011312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stehling, O.; Wilbrecht, C.; Lill, R. Mitochondrial iron–sulfur protein biogenesis and human disease. Biochimie 2014, 100, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Kafina, M.D.; Paw, B.H. Intracellular iron and heme trafficking and metabolism in developing erythroblasts. Metallomics 2017, 9, 1193–1203. [Google Scholar] [CrossRef]
- Barupala, D.P.; Dzul, S.P.; Riggs-Gelasco, P.J.; Stemmler, T.L. Synthesis, delivery and regulation of eukaryotic heme and Fe–S cluster cofactors. Arch. Biochem. Biophys. 2016, 592, 60–75. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Suzuki, T. Iron–sulfur proteins responsible for RNA modifications. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1853, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martínez-Pastor, M.T. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef] [Green Version]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability. Biochim. Biophys. Acta (BBA) 2015, 1853, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, K.; Kaneko, K.; Vargas, P.D. Heme as a Magnificent Molecule with Multiple Missions: Heme Determines Its Own Fate and Governs Cellular Homeostasis. Tohoku J. Exp. Med. 2007, 213, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; De Henau, S.; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; A Nicolet-Dit-Félix, A.; Williams, E.G.; Jha, P.; Sasso, G.L.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, R.J.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exner, N.; Treske, B.; Paquet, D.; Holmstrom, K.; Schiesling, C.; Gispert, S.; Carballo-Carbajal, I.; Berg, D.; Hoepken, H.-H.; Gasser, T.; et al. Loss-of-Function of Human PINK1 Results in Mitochondrial Pathology and Can Be Rescued by Parkin. J. Neurosci. 2007, 27, 12413–12418. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Tanaka, K. Uncovering the roles of PINK1 and parkin in mitophagy. Autophagy 2010, 6, 952–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoepken, H.-H.; Gispert, S.; Morales, B.; Wingerter, O.; Del Turco, D.; Mülsch, A.; Nussbaum, R.L.; Müller, K.; Dröse, S.; Brandt, U.; et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol. Dis. 2007, 25, 401–411. [Google Scholar] [CrossRef]
- Parganlija, D.; Klinkenberg, M.; Domínguez-Bautista, J.; Hetzel, M.; Gispert, S.; Chimi, M.A.; Dröse, S.; Mai, S.; Brandt, U.; Auburger, G.; et al. Loss of PINK1 Impairs Stress-Induced Autophagy and Cell Survival. PLoS ONE 2014, 9, e95288. [Google Scholar] [CrossRef] [Green Version]
- Klinkenberg, M.; Gispert, S.; Dominguez-Bautista, J.A.; Braun, I.; Auburger, G.; Jendrach, M. Restriction of trophic factors and nutrients induces PARKIN expression. Neurogenetics 2011, 13, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Ricciardi, F.; Kurz, A.; Azizov, M.; Hoepken, H.-H.; Becker, D.; Voos, W.; Leuner, K.; Müller, W.E.; Kudin, A.P.; et al. Parkinson Phenotype in Aged PINK1-Deficient Mice Is Accompanied by Progressive Mitochondrial Dysfunction in Absence of Neurodegeneration. PLoS ONE 2009, 4, e5777. [Google Scholar] [CrossRef]
- Visanji, N.P.; Collingwood, J.F.; Finnegan, M.E.; Tandon, A.; House, E.; Hazrati, L.-N. Iron Deficiency in Parkinsonism: Region-Specific Iron Dysregulation in Parkinson’s Disease and Multiple System Atrophy. J. Park. Dis. 2013, 3, 523–537. [Google Scholar] [CrossRef] [Green Version]
- Ndayisaba, A.; Kaindlstorfer, C.; Wenning, G.K. Iron in Neurodegeneration—Cause or Consequence? Front. Neurosci. 2019, 13, 180. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Zhuang, Q.-Q.; Zhu, L.-B.; Zhu, H.; Li, T.; Li, R.; Chen, S.-F.; Huang, C.-P.; Zhang, X.; Zhu, J.-H. Meta-analysis of brain iron levels of Parkinson’s disease patients determined by postmortem and MRI measurements. Sci. Rep. 2016, 6, 36669. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Stroppolo, A.; Gatti, A.; Tampellini, D.; Toscani, M.; Gallorini, M.; Giaveri, G.; Arosio, P.; Santambrogio, P.; Fariello, R.G.; et al. The role of iron and copper molecules in the neuronal vulnerability of locus coeruleus and substantia nigra during aging. Proc. Natl. Acad. Sci. USA 2004, 101, 9843–9848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintze, K.J.; Theil, E.C. Cellular regulation and molecular interactions of the ferritins. Cell. Mol. Life Sci. 2006, 63, 591–600. [Google Scholar] [CrossRef]
- You, L.-H.; Li, Z.; Duan, X.; Zhao, B.-L.; Chang, S.-Y.; Shi, Z.-H. Mitochondrial ferritin suppresses MPTP-induced cell damage by regulating iron metabolism and attenuating oxidative stress. Brain Res. 2016, 1642, 33–42. [Google Scholar] [CrossRef]
- Schweitzer, K.J.; Brüssel, T.; Leitner, P.; Krüger, R.; Bauer, P.; Woitalla, D.; Tomiuk, J.; Gasser, T.; Berg, D. Transcranial ultrasound in different monogenetic subtypes of Parkinson’s disease. J. Neurol. 2007, 254, 613–616. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Cheng, X.; Yuan, H.; Zhu, S.; Liu, J.; Wen, Q.; Xie, Y.; Liu, J.; Kroemer, G.; et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev. Cell 2018, 46, 441–455.e8. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Xie, Y.; Zeh, H.J.; Klionsky, D.J.; Tang, D. Mitochondrial quality control mediated by PINK1 and PRKN: Links to iron metabolism and tumor immunity. Autophagy 2018, 15, 172–173. [Google Scholar] [CrossRef] [Green Version]
- Allen, G.F.; Toth, R.; James, J.; Gangley, I.G. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013, 14, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Vos, M.; Vilain, S.; Swerts, J.; Valadas, J.S.; Van Meensel, S.; Schaap, O.; Verstreken, P. Aconitase Causes Iron Toxicity in Drosophila pink1 Mutants. PLoS Genet. 2013, 9, e1003478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auburger, G.; Gispert, S.; Brehm, N. Methyl-Arginine Profile of Brain from Aged PINK1-KO+A53T-SNCA Mice Suggests Altered Mitochondrial Biogenesis. Park. Dis. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auburger, G.; Gispert, S.; Torres-Odio, S.; Jendrach, M.; Brehm, N.; Canet-Pons, J.; Key, J.; Sen, N.-E. SerThr-PhosphoProteome of Brain from Aged PINK1-KO+A53T-SNCA Mice Reveals pT1928-MAP1B and pS3781-ANK2 Deficits, as Hub between Autophagy and Synapse Changes. Int. J. Mol. Sci. 2019, 20, 3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Odio, S.; Key, J.; Hoepken, H.-H.; Canet-Pons, J.; Valek, L.; Roller, B.; Walter, M.; Morales-Gordo, B.; Meierhofer, D.; Harter, P.N.; et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J. Neuroinflamm. 2017, 14, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gispert, S.; Brehm, N.; Weil, J.; Seidel, K.; Rüb, U.; Kern, B.; Walter, M.; Roeper, J.; Auburger, G. Potentiation of neurotoxicity in double-mutant mice with Pink1 ablation and A53T-SNCA overexpression. Hum. Mol. Genet. 2015, 24, 1061–1076. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Huang, C.-H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [Green Version]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.-M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.-J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(−/−) mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Oelhafen, S.; Hua, H.; Georgiev, O.; Schaffner, W.; Büeler, H. Extended lifespan of Drosophila parkin mutants through sequestration of redox-active metals and enhancement of anti-oxidative pathways. Neurobiol. Dis. 2010, 40, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.F.; Machiela, E.; Dues, D.J.; Spielbauer, K.K.; Senchuk, M.M.; Van Raamsdonk, J.M. Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci. Rep. 2017, 7, 16441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, S.; Yoshimori, T. Autophagy and Longevity. Mol. Cells 2018, 41, 65–72. [Google Scholar] [PubMed]
- Munkácsy, E.; Rea, S.L. The paradox of mitochondrial dysfunction and extended longevity. Exp. Gerontol. 2014, 56, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, J.A.; Ventura, N.; Johnson, T.E.; Rea, S.L. Long-lived mitochondrial (Mit) mutants of Caenorhabditis elegans utilize a novel metabolism. FASEB J. 2010, 24, 4977–4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, F.; Weil, A.; Hamann, A.; Osiewacz, H.D. Human CLPP reverts the longevity phenotype of a fungal ClpP deletion strain. Nat. Commun. 2013, 4, 1397. [Google Scholar] [CrossRef] [Green Version]
- Gispert, S.; Parganlija, D.; Klinkenberg, M.; Dröse, S.; Wittig, I.; Mittelbronn, M.; Grzmil, P.; Koob, S.; Hamann, A.; Walter, M.; et al. Loss of mitochondrial peptidase Clpp leads to infertility, hearing loss plus growth retardation via accumulation of CLPX, mtDNA and inflammatory factors. Hum. Mol. Genet. 2013, 22, 4871–4887. [Google Scholar] [CrossRef] [Green Version]
- Bhaskaran, S.; Pharaoh, G.; Ranjit, R.; Murphy, A.; Matsuzaki, S.; Nair, B.C.; Forbes, B.; Gispert, S.; Auburger, G.; Humphries, K.M.; et al. Loss of mitochondrial protease ClpP protects mice from diet-induced obesity and insulin resistance. EMBO Rep. 2018, 19, e45009. [Google Scholar] [CrossRef]
- Mai, S.; Klinkenberg, M.; Auburger, G.; Bereiter-Hahn, J.; Jendrach, M. Decreased expression of Drp1 and Fis1 mediates mitochondrial elongation in senescent cells and enhances resistance to oxidative stress through PINK1. J. Cell Sci. 2010, 123, 917–926. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.H.; Rivadeneira, D.B.; Caino, M.C.; Chae, Y.C.; Speicher, D.W.; Tang, H.-Y.; Vaira, V.; Bosari, S.; Palleschi, A.; Rampini, P.; et al. The Mitochondrial Unfoldase-Peptidase Complex ClpXP Controls Bioenergetics Stress and Metastasis. PLoS Biol. 2016, 14, e1002507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexeyev, M.F. Is there more to aging than mitochondrial DNA and reactive oxygen species? FEBS J. 2009, 276, 5768–5787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, A.B.; Jeong, D.-E.; Lee, S.-J.V. Mitochondria and Organismal Longevity. Curr. Genom. 2012, 13, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.T.; Liu, X.; Ye, C.Y.; Tao, L.X.; Zhou, H.; Zhang, H.-Y. Iron-induced energy supply deficiency and mitochondrial fragmentation in neurons. J. Neurochem. 2018, 147, 816–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2018, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Gallardo, A.K.; Missirlis, F. Cellular iron sensing and regulation: Nuclear IRP1 extends a classic paradigm. Biochim. Biophys. Acta (BBA) 2020, 1867, 118705. [Google Scholar] [CrossRef]
- Camaschella, C.; Campanella, A.; De Falco, L.; Boschetto, L.; Merlini, R.; Silvestri, L.; Levi, S.; Iolascon, A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood 2007, 110, 1353–1358. [Google Scholar] [CrossRef]
- Lipper, C.H.; Paddock, M.L.; Onuchic, J.N.; Mittler, R.; Nechushtai, R.; Jennings, P.A. Cancer-Related NEET Proteins Transfer 2Fe-2S Clusters to Anamorsin, a Protein Required for Cytosolic Iron-Sulfur Cluster Biogenesis. PLoS ONE 2015, 10, e0139699. [Google Scholar] [CrossRef] [Green Version]
- Mons, C.; Ferecatu, I.; Riquier, S.; Lescop, E.; Bouton, C.; Golinelli, M.-P. Combined Biochemical, Biophysical, and Cellular Methods to Study Fe–S Cluster Transfer and Cytosolic Aconitase Repair by MitoNEET. Methods Enzymol. 2017, 595, 83–106. [Google Scholar] [CrossRef]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Long, D. Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 2020, 14, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, D.M.; Jones, H.B.L.; Brown, D.R. Levels of alpha- and beta-synuclein regulate cellular susceptibility to toxicity from alpha-synuclein oligomers. FASEB J. 2018, 32, 995–1006. [Google Scholar] [CrossRef] [Green Version]
- Su, B.; Liu, H.; Wang, X.; Chen, S.G.; Siedlak, S.L.; Kondo, E.; Choi, R.; Takeda, A.; Castellani, R.J.; Perry, G.; et al. Ectopic localization of FOXO3a protein in Lewy bodies in Lewy body dementia and Parkinson’s disease. Mol. Neurodegener. 2009, 4, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, K.; Ispierto, L.; Latorre, P.; Tolosa, E.; Ariza, A. Alpha- and beta-synuclein expression in Parkinson disease with and without dementia. J. Neurol. Sci. 2011, 310, 112–117. [Google Scholar] [CrossRef]
- Brighina, L.; Okubadejo, N.U.; Schneider, N.K.; Lesnick, T.G.; De Andrade, M.; Cunningham, J.M.; Farrer, M.J.; Lincoln, S.J.; Rocca, W.A.; Maraganore, D.M. Beta-synuclein gene variants and Parkinson’s disease: A preliminary case-control study. Neurosci. Lett. 2007, 420, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Davies, P.; Moualla, D.; Brown, D.R. Alpha-Synuclein Is a Cellular Ferrireductase. PLoS ONE 2011, 6, e15814. [Google Scholar] [CrossRef]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, R.; Verbitsky, M.; Kisselev, S.; Browne, A.; Mejia-Sanatana, H.; Louis, E.D.; Cote, L.J.; Andrews, H.F.; Waters, C.H.; et al. Genome-Wide association study identifies candidate genes for Parkinson’s disease in an Ashkenazi Jewish population. BMC Med Genet. 2011, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahut, S.; Gispert, S.; Ömür, Ö.; Depboylu, C.; Seidel, K.; Domínguez-Bautista, J.A.; Brehm, N.; Tireli, H.; Hackmann, K.; Pirkevi, C.; et al. Blood RNA biomarkers in prodromal PARK4 and rapid eye movement sleep behavior disorder show role of complexin 1 loss for risk of Parkinson’s disease. Dis. Model. Mech. 2017, 10, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Li, C.; Lv, S.; Zhou, B. Pantothenate kinase-associated neurodegeneration: Insights from a Drosophila model. Hum. Mol. Genet. 2009, 18, 3659–3672. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Kanthasamy, A.G.; Anantharam, V.; Sun, F.; Kanthasamy, A.G. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: Relevance to epigenetic mechanisms of neurodegeneration. Mol. Pharmacol. 2010, 77, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta (BBA) 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Lanford, R.; Mueller, S.; Gerhard, G.S.; Luscieti, S.; Sanchez, M.; Devireddy, L. Siderophore-mediated iron trafficking in humans is regulated by iron. J. Mol. Med. 2012, 90, 1209–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Yang, S.; Tan, G.; Ye, W.; Liu, D.; Qian, X.; Ding, Z.; Zhong, Y.; Zhang, J.; Jiang, D.; et al. Reduction of Mitoferrin Results in Abnormal Development and Extended Lifespan in Caenorhabditis elegans. PLoS ONE 2012, 7, e29666. [Google Scholar] [CrossRef] [Green Version]
- Yanatori, I.; Richardson, D.R.; Toyokuni, S.; Kishi, F. The iron chaperone poly(rC)-binding protein 2 forms a metabolon with the heme oxygenase 1/cytochrome P450 reductase complex for heme catabolism and iron transfer. J. Biol. Chem. 2017, 292, 13205–13229. [Google Scholar] [CrossRef] [Green Version]
- Leidgens, S.; Bullough, K.Z.; Shi, H.; Li, F.; Shakoury-Elizeh, M.; Yabe-Wada, T.; Subramanian, P.; Hsu, E.; Natarajan, N.; Nandal, A.; et al. Each Member of the Poly-r(C)-binding Protein 1 (PCBP) Family Exhibits Iron Chaperone Activity toward Ferritin. J. Biol. Chem. 2013, 288, 17791–17802. [Google Scholar] [CrossRef] [Green Version]
- Galy, B.; Ferring-Appel, D.; Sauer, S.W.; Kaden, S.; Lyoumi, S.; Puy, H.; Kölker, S.; Gröne, H.-J.; Hentze, M.W. Iron Regulatory Proteins Secure Mitochondrial Iron Sufficiency and Function. Cell Metab. 2010, 12, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhao, H.; Hao, S.; Shang, L.; Wu, J.; Song, C.; Meyron-Holtz, E.G.; Qiao, T.; Li, K. Iron regulatory protein deficiency compromises mitochondrial function in murine embryonic fibroblasts. Sci. Rep. 2018, 8, 5118. [Google Scholar] [CrossRef]
- Chen, W.; Dailey, H.A.; Paw, B.H. Ferrochelatase forms an oligomeric complex with mitoferrin-1 and Abcb10 for erythroid heme biosynthesis. Blood 2010, 116, 628–630. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Bayeva, M.; Ghanefar, M.; Potini, V.; Sun, L.; Mutharasan, R.K.; Wu, R.; Khechaduri, A.; Naik, T.J.; Ardehali, H. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc. Natl. Acad. Sci. USA 2012, 109, 4152–4157. [Google Scholar] [CrossRef] [Green Version]
- Pondarré, C.; Antiochos, B.B.; Campagna, D.R.; Clarke, S.L.; Greer, E.L.; Deck, K.M.; McDonald, A.; Han, A.-P.; Medlock, A.; Kutok, J.L.; et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron–sulfur cluster biogenesis. Hum. Mol. Genet. 2006, 15, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R.; Dutkiewicz, R.; Freibert, S.A.; Heidenreich, T.; Mascarenhas, J.; Netz, D.J.; Paul, V.D.; Pierik, A.J.; Richter, N.; Stümpfig, M.; et al. The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron–sulfur proteins. Eur. J. Cell Biol. 2015, 94, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Kim, K.S.; Holmes-Hampton, G.; Singh, A.; Rouault, T.A. Dimeric ferrochelatase bridges ABCB7 and ABCB10 homodimers in an architecturally defined molecular complex required for heme biosynthesis. Haematogica. 2019, 104, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Mühlenhoff, U.; Hoffmann, B.; Richter, N.; Rietzschel, N.; Spantgar, F.; Stehling, O.; Uzarska, M.A.; Lill, R. Compartmentalization of iron between mitochondria and the cytosol and its regulation. Eur. J. Cell Biol. 2015, 94, 292–308. [Google Scholar] [CrossRef]
- Maio, N.; Rouault, T.A. Mammalian Fe–S proteins: Definition of a consensus motif recognized by the co-chaperone HSC20. Metallomics 2016, 8, 1032–1046. [Google Scholar] [CrossRef] [Green Version]
- Uzarska, M.A.; Nasta, V.; Weiler, B.D.; Spantgar, F.; Ciofi-Baffoni, S.; Saviello, M.R.; Gonnelli, L.; Mühlenhoff, U.; Banci, L.; Lill, R. Mitochondrial Bol1 and Bol3 function as assembly factors for specific iron-sulfur proteins. eLife 2016, 5, 83. [Google Scholar] [CrossRef]
- Braymer, J.J.; Lill, R. Iron–sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [Green Version]
- Choi, A.M.; Alam, J. Heme oxygenase-1: Function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef]
- Mast, N.; Charvet, C.; Pikuleva, I.A.; Stout, C.D. Structural Basis of Drug Binding to CYP46A1, an Enzyme That Controls Cholesterol Turnover in the Brain. J. Biol. Chem. 2010, 285, 31783–31795. [Google Scholar] [CrossRef] [Green Version]
- Correia, M.A.; Sinclair, P.R.; De Matteis, F. Cytochrome P450 regulation: The interplay between its heme and apoprotein moieties in synthesis, assembly, repair, and disposal. Drug Metab. Rev. 2011, 43, 1–26. [Google Scholar] [CrossRef] [Green Version]
- De Sanctis, D.; Dewilde, S.; Pesce, A.; Moens, L.; Ascenzi, P.; Hankeln, T.; Burmester, T.; Bolognesi, M. Crystal Structure of Cytoglobin: The Fourth Globin Type Discovered in Man Displays Heme Hexa-coordination. J. Mol. Biol. 2004, 336, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Barthelme, M.; Scheele, U.; Dinkelaker, S.; Janoschka, A.; Macmillan, F.; Albers, S.-V.; Driessen, A.J.M.; Stagni, M.S.; Bill, E.; Meyer-Klaucke, W.; et al. Structural Organization of Essential Iron-Sulfur Clusters in the Evolutionarily Highly Conserved ATP-binding Cassette Protein ABCE1. J. Biol. Chem. 2007, 282, 14598–14607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhebshi, A.; Sideri, T.C.; Holland, S.L.; Avery, S.V. The essential iron-sulfur protein Rli1 is an important target accounting for inhibition of cell growth by reactive oxygen species. Mol. Biol. Cell 2012, 23, 3582–3590. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Johansson, M.J.; Byström, A.S. An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 2005, 11, 424–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta (BBA) 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbirt, K.K.; Bonkovsky, H.L. Heme oxygenase: Recent advances in understanding its regulation and role. Proc. Assoc. Am. Phys. 1999, 111, 438–447. [Google Scholar] [CrossRef]
- Chung, J.; Anderson, S.; Gwynn, B.; Deck, K.M. Iron regulatory protein-1 protects against mitoferrin-1-deficient porphyria. J. Biol. Chem. 2014, 289, 7835–7843. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Sun, X.; Wang, L.; Wong, Y.K.; Lee, Y.M.; Zhou, C.; Wu, G.; Zhao, T.; Yang, L.; Lu, L.; et al. Artesunate-induced mitophagy alters cellular redox status. Redox Biol. 2018, 19, 263–273. [Google Scholar] [CrossRef]
- Salvador, G.A. Iron in neuronal function and dysfunction. BioFactors 2010, 36, 103–110. [Google Scholar] [CrossRef]
- Hirsch, E.C. Altered regulation of iron transport and storage in Parkinson’s disease. J. Neural. Transm. Suppl. 2006, 71, 201–204. [Google Scholar] [CrossRef]
- Dexter, D.T.; Carayon, A.; Vidailhet, M.; Ruberg, M.; Agid, F.; Agid, Y.; Lees, A.J.; Wells, F.R.; Jenner, P.; Marsden, C.D. Decreased Ferritin Levels in Brain in Parkinson’s Disease. J. Neurochem. 1990, 55, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Bauckman, K.A.; Haller, E.M.; Flores, I.; Nanjundan, M. Iron modulates cell survival in a Ras- and MAPK-dependent manner in ovarian cells. Cell Death Dis. 2013, 4, e592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.-J.; Tu, H.-J.; Chien, W.-L.; Kang, K.-H.; Lu, D.-H.; Liou, H.-H.; Lee, M.-J.; Fu, W.-M. Antagonism of proteasome inhibitor-induced heme oxygenase-1 expression by PINK1 mutation. PLoS ONE 2017, 12, e0183076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requejo, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolaños, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.-H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yan, J.; Sun, C.; Li, G.; Li, S.; Zhang, L.; Di, C.; Gan, L.; Wang, Y.; Zhou, R.; et al. Ameliorating mitochondrial dysfunction restores carbon ion-induced cognitive deficits via co-activation of NRF2 and PINK1 signaling pathway. Redox Biol. 2018, 17, 143–157. [Google Scholar] [CrossRef]
- Chen, X.-L.; Kunsch, C. Induction of cytoprotective genes through Nrf2/antioxidant response element pathway: A new therapeutic approach for the treatment of inflammatory diseases. Curr. Pharm. Des. 2004, 10, 879–891. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. The Roles of NRF2 in Modulating Cellular Iron Homeostasis. Antioxidants Redox Signal. 2018, 29, 1756–1773. [Google Scholar] [CrossRef] [Green Version]
- Lau, A.; Wang, X.-J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xiang, Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G. Proline hydroxylation and gene expression. Annu. Rev. Biochem. 2005, 74, 115–128. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.-A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Huynh, N.; Ou, Q.; Cox, P.; Lill, R.; King-Jones, K. Glycogen branching enzyme controls cellular iron homeostasis via Iron Regulatory Protein 1 and mitoNEET. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Kessler, K.R.; Hamscho, N.; Morales, B.; Menzel, C.; Barrero, F.; Vives, F.; Gispert, S.; Auburger, G. Dopaminergic function in a family with the PARK6 form of autosomal recessive Parkinson’s syndrome. J. Neural Transm. 2005, 112, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Tuin, I.; Voss, U.; Kessler, K.; Krakow, K.; Hilker, R.; Morales, B.; Steinmetz, H.; Auburger, G. Sleep quality in a family with hereditary parkinsonism (PARK6). Sleep Med. 2008, 9, 684–688. [Google Scholar] [CrossRef]
- Hoepken, H.-H.; Gispert, S.; Azizov, M.; Klinkenberg, M.; Ricciardi, F.; Kurz, A.; Morales-Gordo, B.; Bonin, M.; Riess, O.; Gasser, T.; et al. Parkinson patient fibroblasts show increased alpha-synuclein expression. Exp. Neurol. 2008, 212, 307–313. [Google Scholar] [CrossRef]
- Klinkenberg, M.; Thurow, N.; Gispert, S.; Ricciardi, F.; Eich, F.; Prehn, J.H.M.; Auburger, G.; Kögel, D. Enhanced vulnerability of PARK6 patient skin fibroblasts to apoptosis induced by proteasomal stress. Neuroscience 2010, 166, 422–434. [Google Scholar] [CrossRef]
- Auburger, G.; Klinkenberg, M.; Drost, J.; Marcus, K.; Morales-Gordo, B.; Kunz, W.S.; Brandt, U.; Broccoli, V.; Reichmann, H.; Gispert, S.; et al. Primary Skin Fibroblasts as a Model of Parkinson’s Disease. Mol. Neurobiol. 2012, 46, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Gehrke, S.; Wu, Z.; Klinkenberg, M.; Sun, Y.; Auburger, G.; Guo, S.; Lu, B. PINK1 and Parkin control localized translation of respiratory chain component mRNAs on mitochondria outer membrane. Cell Metab. 2015, 21, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Azkona, G.; De Maturana, R.L.; Del Rio, P.; Sousa, A.; Vazquez, N.; Zubiarrain, A.; Jimenez-Blasco, D.; Bolaños, J.P.; Morales, B.; Auburger, G.; et al. LRRK2 Expression Is Deregulated in Fibroblasts and Neurons from Parkinson Patients with Mutations in PINK1. Mol. Neurobiol. 2018, 55, 506–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Tantray, I.; Lim, J.; Chen, S.; Li, Y.; Davis, Z.; Sitron, C.; Dong, J.; Gispert, S.; Auburger, G.; et al. MISTERMINATE Mechanistically Links Mitochondrial Dysfunction with Proteostasis Failure. Mol. Cell 2019, 75, 835–848.e8. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R. alpha-Synuclein as a ferrireductase. Biochem. Soc. Trans. 2013, 41, 1513–1517. [Google Scholar] [PubMed] [Green Version]
- McDowall, J.S.; Ntai, I.; Honeychurch, K.C.; Hart, J.P.; Colin, P.; Schneider, B.; Brown, D.R. Alpha-synuclein ferrireductase activity is detectible in vivo, is altered in Parkinson’s disease and increases the neurotoxicity of DOPAL. Mol. Cell. Neurosci. 2017, 85, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDowall, J.S.; Brown, D.R. Alpha-synuclein: Relating metals to structure, function and inhibition. Metallomics 2016, 8, 385–397. [Google Scholar] [CrossRef] [PubMed]
- McLeary, F.; Rcom-H’Cheo-Gauthier, A.N.; Goulding, M.; Radford, R.; Okita, Y.; Faller, P.; Chung, R.S.; Pountney, D.L. Switching on Endogenous Metal Binding Proteins in Parkinson’s Disease. Cells 2019, 8, 179. [Google Scholar] [CrossRef] [Green Version]
- Roberts, H.L.; Schneider, B.L.; Brown, D.R. alpha-Synuclein increases beta-amyloid secretion by promoting beta-/gamma-secretase processing of APP. PLoS ONE 2017, 12, e0171925. [Google Scholar]
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for α-synuclein in Parkinson’s disease: Alteration of ER-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033. [Google Scholar] [CrossRef]
- Wolozin, B.; Golts, N. Book Review: Iron and Parkinson’s Disease. Neuroscientist 2002, 8, 22–32. [Google Scholar] [CrossRef]
- Chen, B.; Wen, X.; Jiang, H.; Wang, J.; Song, N.; Xie, J. Interactions between iron and α-synuclein pathology in Parkinson’s disease. Free Radic. Biol. Med. 2019, 141, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Naitoh, Y.; Kohno, H.; Tokunaga, R.; Taketani, S. Iron deprivation decreases ribonucleotide reductase activity and DNA synthesis. Life Sci. 1992, 50, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Erlitzki, R.; Long, J.C.; Theil, E.C. Multiple, conserved iron-responsive elements in the 3′-untranslated region of transferrin receptor mRNA enhance binding of iron regulatory protein 2. J. Biol. Chem. 2002, 277, 42579–42587. [Google Scholar]
- Li, J.; Braganza, A.; Sobol, R.W. Base Excision Repair Facilitates a Functional Relationship Between Guanine Oxidation and Histone Demethylation. Antioxid. Redox Signal. 2013, 18, 2429–2443. [Google Scholar] [CrossRef]
- Martelli, A.; Wattenhofer-Donzé, M.; Schmucker, S.; Bouvet, S.; Reutenauer, L.; Puccio, H. Frataxin is essential for extramitochondrial Fe–S cluster proteins in mammalian tissues. Hum. Mol. Genet. 2007, 16, 2651–2658. [Google Scholar] [CrossRef]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.-M.; Linehan, W.M.; Rouault, T.A. Acute loss of iron–sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [Green Version]
- Haskamp, V.; Karrie, S.; Mingers, T.; Barthels, S.; Alberge, F.; Magalon, A.; Müller, K.; Bill, E.; Lubitz, W.; Kleeberg, K.; et al. The radical SAM protein HemW is a heme chaperone. J. Biol. Chem. 2017, 293, 2558–2572. [Google Scholar] [CrossRef] [Green Version]
- Latorre-Pellicer, A.; Moreno-Loshuertos, R.; Lechuga-Vieco, A.V.; Sánchez-Cabo, F.; Torroja, C.; Acín-Pérez, R.; Calvo, E.; Aix, E.; González-Guerra, A.; Logan, A.; et al. Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 2016, 535, 561–565. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington’s Disease. Cell 2015, 162, 516–526. [Google Scholar]
- Jones, L.; Houlden, H.; Tabrizi, S.J. DNA repair in the trinucleotide repeat disorders. Lancet Neurol. 2017, 16, 88–96. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J.; Caldecott, K.W. DNA Strand Break Repair and Human Genetic Disease. Annu. Rev. Genom. Hum. Genet. 2007, 8, 37–55. [Google Scholar] [CrossRef]
- Piel, R.B.; Shiferaw, M.T.; Vashisht, A.A.; Marcero, J.R.; Praissman, J.L.; Phillips, J.D.; Wohlschlegel, J.A.; Medlock, A. A Novel Role for Progesterone Receptor Membrane Component 1 (PGRMC1): A Partner and Regulator of Ferrochelatase. Biochemistry 2016, 55, 5204–5217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Kim, S.K.; Pai, S.H. Changes in serum lipid concentrations during iron depletion and after iron supplementation. Ann. Clin. Lab. Sci. 2001, 31, 151–156. [Google Scholar]
- Llorens, J.V.; Metzendorf, C.; Missirlis, F.; Lind, M.I. Mitochondrial iron supply is required for the developmental pulse of ecdysone biosynthesis that initiates metamorphosis in Drosophila melanogaster. JBIC J. Biol. Inorg. Chem. 2015, 20, 1229–1238. [Google Scholar] [CrossRef]
- Wu, Z.; Wang, Y.; Lim, J.; Liu, B.; Li, Y.; Vartak, R.; Stankiewicz, T.; Montgomery, S.; Lu, B. Ubiquitination of ABCE1 by NOT4 in Response to Mitochondrial Damage Links Co-translational Quality Control to PINK1-Directed Mitophagy. Cell Metab. 2018, 28, 130–144. [Google Scholar]
- Mehta, R.; Chandler-Brown, D.; Ramos, F.J.; Shamieh, L.S.; Kaeberlein, M. Regulation of mRNA Translation as a Conserved Mechanism of Longevity Control. Adv. Exp. Med. Biol. 2010, 694, 14–29. [Google Scholar] [CrossRef]
- MacInnes, A.W. The role of the ribosome in the regulation of longevity and lifespan extension. Wiley Interdiscip. Rev. RNA 2016, 7, 198–212. [Google Scholar] [CrossRef]
- Coelho, C.M.A.; Kolevski, B.; Bunsn, C.; Walker, C.; Dahanukar, A.; Leevers, S.J. Growth and cell survival are unevenly impaired in pixie mutant wing discs. Development 2005, 132, 5411–5424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nürenberg-Goloub, E.; Heinemann, H.; Gerovac, M.; Tampé, R. Ribosome recycling is coordinated by processive events in two asymmetric ATP sites of ABCE1. Life Sci. Alliance 2018, 1, e201800095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Lee, H.; Dominguez, D.; Heiman, M.; Burge, C.B. Widespread Accumulation of Ribosome-Associated Isolated 3′ UTRs in Neuronal Cell Populations of the Aging Brain. Cell Rep. 2018, 25, 2447–2456.e4. [Google Scholar] [PubMed] [Green Version]
- Le Roy, F.; Bisbal, C.; Silhol, M.; Martinand, C.; Lebleu, B.; Salehzada, T. The 2-5A/RNase L/RNase L inhibitor (RLI) [correction of (RNI)] pathway regulates mitochondrial mRNAs stability in interferon alpha-treated H9 cells. J. Biol. Chem. 2001, 276, 48473–48482. [Google Scholar]
- Stadhouders, R.; Aktuna, S.; Thongjuea, S.; Aghajanirefah, A.; Pourfarzad, F.; Van Ijcken, W.; Lenhard, B.; Rooks, H.; Best, S.; Menzel, S.; et al. HBS1L-MYB intergenic variants modulate fetal hemoglobin via long-range MYB enhancers. J. Clin. Investig. 2014, 124, 1699–1710. [Google Scholar] [CrossRef] [Green Version]
- Volani, C.; Paglia, G.; Smarason, S.V.; Pramstaller, P.P.; Demetz, E.; Pfeifhofer-Obermair, C.; Weiss, G. Metabolic Signature of Dietary Iron Overload in a Mouse Model. Cells 2018, 7, 264. [Google Scholar] [CrossRef] [Green Version]
- Finazzi, D.; Arosio, P. Biology of ferritin in mammals: An update on iron storage, oxidative damage and neurodegeneration. Arch. Toxicol. 2014, 88, 1787–1802. [Google Scholar] [CrossRef] [Green Version]
- Yévenes, A. The Ferritin Superfamily. Plant Microbe Interact. 2017, 83, 75–102. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Benkovic, S.A.; Lin, L.; Yonutas, H.M.; Crish, S.D.; Sullivan, P.G.; Darvesh, A.S.; Brown, C.M.; Richardson, J.R. MitoNEET (CISD1) Knockout Mice Show Signs of Striatal Mitochondrial Dysfunction and a Parkinson’s Disease Phenotype. ACS Chem. Neurosci. 2017, 8, 2759–2765. [Google Scholar] [CrossRef]
- Lee, J.; You, J.H.; Shin, D.; Roh, J.-L. Inhibition of Glutaredoxin 5 predisposes Cisplatin-resistant Head and Neck Cancer Cells to Ferroptosis. Theranostics 2020, 10, 7775–7786. [Google Scholar] [CrossRef]
- Chi, H.; Tang, W.; Bai, Y. Molecular evidence of impaired iron metabolism and its association with Parkinson’s disease progression. 3 Biotech 2020, 10, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Faucheux, B.A.; Martin, M.-E.; Beaumont, C.; Hunot, S.; Hauw, J.-J.; Agid, Y.; Hirsch, E.C. Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J. Neurochem. 2002, 83, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Núñez, M.T. Iron dyshomeostasis in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 71, 205–213. [Google Scholar] [CrossRef]
- Mena, N.P.; Bulteau, A.-L.; Salazar, J.; Hirsch, E.C.; Núñez, M.T. Effect of mitochondrial complex I inhibition on Fe–S cluster protein activity. Biochem. Biophys. Res. Commun. 2011, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Song, N.; Guo, X.; Jiang, H.; Zhang, H.; Xie, J. Differences in vulnerability of neurons and astrocytes to heme oxygenase-1 modulation: Implications for mitochondrial ferritin. Sci. Rep. 2016, 6, 24200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, W.; Ikematsu, K.; Tsuda, R. Age-associated increases in heme oxygenase-1 and ferritin immunoreactivity in the autopsied brain. Leg. Med. 2003, 5 (Suppl. 1), S360–S366. [Google Scholar] [CrossRef]
- Galazka-Friedman, J.; Bauminger, E.R.; Koziorowski, D.; Friedman, A. Mössbauer spectroscopy and ELISA studies reveal differences between Parkinson’s disease and control substantia nigra. Biochim. Biophys. Acta (BBA) 2004, 1688, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Wypijewska, A.; Galazka-Friedman, J.; Bauminger, E.R.; Wszolek, Z.K.; Schweitzer, K.J.; Dickson, D.W.; Jaklewicz, A.; Elbaum, D.; Friedman, A. Iron and reactive oxygen species activity in parkinsonian substantia nigra. Park. Relat. Disord. 2010, 16, 329–333. [Google Scholar] [CrossRef]
- Huo, L.-R.; Shen, C.; Ju, W.; Zou, J.-H.; Yan, W.; Brown, W.T.; Zhong, N. Identification of novel partner proteins of PCBP1. J. Peking Univ. Heal. Sci. 2009, 41, 402–408. [Google Scholar]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A Cytosolic Iron Chaperone That Delivers Iron to Ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef] [Green Version]
- Henn, I.H.; Bouman, B.; Schlehe, J.S.; Schlierf, A.; Schramm, J.E.; Wegener, E.; Nakaso, K.; Culmsee, C.; Berninger, B.; Krappmann, D.; et al. Parkin mediates neuroprotection through activation of IkappaB kinase/nuclear factor-kappaB signaling. J. Neurosci. 2007, 27, 1868–1878. [Google Scholar] [PubMed] [Green Version]
- Williams, N.C.; O’Neill, L.A. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen-Roetling, J.; Liu, W.; Regan, R.F. Iron accumulation and neurotoxicity in cortical cultures treated with holotransferrin. Free Radic. Biol. Med. 2011, 51, 1966–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avila, F.; Echeverría, G.; Pérez, D.; Martinez, C.; Strobel, P.; Castillo, O.; Villaroel, L.; Mezzano, D.; Rozowski, J.; Urquiaga, I.; et al. Serum Ferritin Is Associated with Metabolic Syndrome and Red Meat Consumption. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Lane, D.J.R.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef]
- Reilly, D.K.; Hershey, L.; Rivera-Calimlim, L.; Shoulson, I. On-off effects in Parkinson’s disease: A controlled investigation of ascorbic acid therapy. Adv. Neurol. 1983, 37, 51–60. [Google Scholar]
- Yapa, S.S. Detection of subclinical ascorbate deficiency in early Parkinson’s disease. Public Health 1992, 106, 393–395. [Google Scholar] [CrossRef]
- Fahn, S. A pilot trial of high-dose alpha-tocopherol and ascorbate in early Parkinson’s disease. Ann. Neurol. 1992, 32, S128–S132. [Google Scholar] [CrossRef]
- Logroscino, G.; Gao, X.; Chen, H.; Wing, A.; Ascherio, A. Dietary Iron Intake and Risk of Parkinson’s Disease. Am. J. Epidemiol. 2008, 168, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Wolk, A.; Håkansson, N.; Pedersen, N.L.; Wirdefeldt, K. Dietary antioxidants and risk of Parkinson’s disease in two population-based cohorts. Mov. Disord. 2017, 32, 1631–1636. [Google Scholar] [CrossRef]
- Wan, Z.; Xu, J.; Huang, Y.; Zhai, Y.; Ma, Z.; Zhou, B.; Cao, Z. Elevating bioavailable iron levels in mitochondria suppresses the defective phenotypes caused by PINK1 loss-of-function in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2020. [Google Scholar] [CrossRef]
- Seguin, A.; Takahashi-Makise, N.; Yien, Y.Y.; Huston, N.C.; Whitman, J.C.; Musso, G.; Wallace, J.A.; Bradley, T.; Bergonia, H.A.; Kafina, M.D.; et al. Reductions in the mitochondrial ABC transporter Abcb10 affect the transcriptional profile of heme biosynthesis genes. J. Biol. Chem. 2017, 292, 16284–16299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Paradkar, P.N.; Li, L.; Pierce, E.L.; Langer, N.B.; Takahashi-Makise, N.; Hyde, B.B.; Shirihai, O.S.; Ward, D.M.; Kaplan, J.; et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc. Natl. Acad. Sci. USA 2009, 106, 16263–16268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial Iron Import through Differential Turnover of Mitoferrin 1 and Mitoferrin 2. Mol. Cell. Biol. 2008, 29, 1007–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Sen, N.-E.; Canet-Pons, J.; Halbach, M.V.; Arsovic, A.; Pilatus, U.; Chae, W.-H.; Kaya, Z.-E.; Seidel, K.; Rollmann, E.; Mittelbronn, M.; et al. Generation of an Atxn2-CAG100 knock-in mouse reveals N-acetylaspartate production deficit due to early Nat8l dysregulation. Neurobiol. Dis. 2019, 132, 104559. [Google Scholar] [CrossRef]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Gielisch, I.; Meierhofer, D. Metabolome and Proteome Profiling of Complex I Deficiency Induced by Rotenone. J. Proteome Res. 2014, 14, 224–235. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Key, J.; Sen, N.E.; Arsović, A.; Krämer, S.; Hülse, R.; Khan, N.N.; Meierhofer, D.; Gispert, S.; Koepf, G.; Auburger, G. Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells 2020, 9, 2229. https://doi.org/10.3390/cells9102229
Key J, Sen NE, Arsović A, Krämer S, Hülse R, Khan NN, Meierhofer D, Gispert S, Koepf G, Auburger G. Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells. 2020; 9(10):2229. https://doi.org/10.3390/cells9102229
Chicago/Turabian StyleKey, Jana, Nesli Ece Sen, Aleksandar Arsović, Stella Krämer, Robert Hülse, Natasha Nadeem Khan, David Meierhofer, Suzana Gispert, Gabriele Koepf, and Georg Auburger. 2020. "Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence" Cells 9, no. 10: 2229. https://doi.org/10.3390/cells9102229
APA StyleKey, J., Sen, N. E., Arsović, A., Krämer, S., Hülse, R., Khan, N. N., Meierhofer, D., Gispert, S., Koepf, G., & Auburger, G. (2020). Systematic Surveys of Iron Homeostasis Mechanisms Reveal Ferritin Superfamily and Nucleotide Surveillance Regulation to be Modified by PINK1 Absence. Cells, 9(10), 2229. https://doi.org/10.3390/cells9102229