Deletion of Acid-Sensing Ion Channel 3 Relieves the Late Phase of Neuropathic Pain by Preventing Neuron Degeneration and Promoting Neuron Repair

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Surgery
2.3. Histological Staining and Immunostaining
2.4. Tests
2.5. Statistical Analysis
3. Results
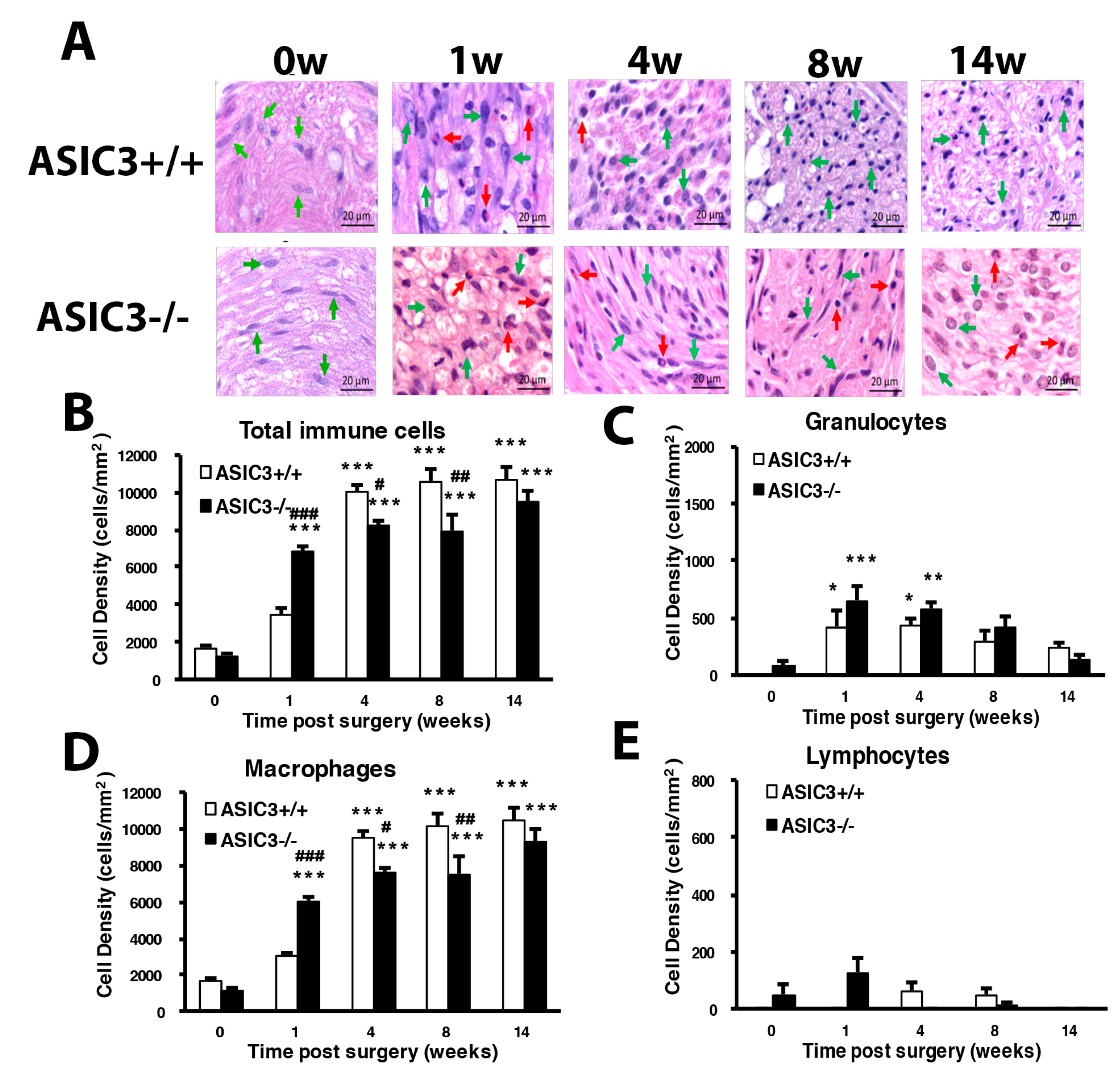
3.1. CCI of Sciatic Nerve Causes Long-Term Hyperalgesia, Inflammation, and Neuron Degeneration
3.2. Number of SGCs and ATF3-Positive Neurons Was Increased after CCI Surgery
3.3. ASIC3 Deficiency Reverses CCI-Induced Hyperalgesia and Delays Neuron Loss in the Beginning of CCI
3.4. ASIC3 Deficiency Reverses the Shift from Large to Small Cells in ATF3+ Neurons, with No Alteration in Gliosis
3.5. ASIC3 Deletion Alters M1/ M2 Macrophage Ratio after CCI
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reichling, D.B.; Levine, J.D. Critical role of nociceptor plasticity in chronic pain. Trends Neurosci. 2009, 32, 611–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, J.; Woolf, C.J. The neuropathic pain triad: Neurons, immune cells and glia. Nat. Neurosci. 2007, 10, 1361–1368. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, B.L.; Campenot, R.B. Regulation of Wallerian degeneration and nerve growth factor withdrawal-induced pruning of axons of sympathetic neurons by the proteasome and the MEK/Erk pathway. Mol. Cell. Neurosci. 2005, 28, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Sacerdote, P.; Franchi, S.; Trovato, A.E.; Valsecchi, A.E.; Panerai, A.E.; Colleoni, M. Transient early expression of TNF-alpha in sciatic nerve and dorsal root ganglia in a mouse model of painful peripheral neuropathy. Neurosci. Lett. 2008, 436, 210–213. [Google Scholar] [CrossRef]
- Shamash, S.; Reichert, F.; Rotshenker, S. The cytokine network of Wallerian degeneration: Tumor necrosis factor-alpha, interleukin-1alpha, and interleukin-1beta. J. Neurosci. 2002, 22, 3052–3060. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.F.; Moalem-Taylor, G. Detailed characterization of neuro-immune responses following neuropathic injury in mice. Brain Res. 2011, 1405, 95–108. [Google Scholar] [CrossRef]
- Mueller, M.; Wacker, K.; Ringelstein, E.B.; Hickey, W.F.; Imai, Y.; Kiefer, R. Rapid response of identified resident endoneurial macrophages to nerve injury. Am. J. Pathol. 2001, 159, 2187–2197. [Google Scholar] [CrossRef]
- Vega-Avelaira, D.; Géranton, S.M.; Fitzgerald, M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain 2009, 5, 70. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.; Attal, N.; Haroutounian, S.; McNicol, E.; Baron, R.; Dworkin, R.H.; Gilron, I.; Haanpää, M.; Hansson, P.; Jensen, T.S.; et al. Pharmacotherapy for neuropathic pain in adults: A systematic review and meta-analysis. Lancet Neurol. 2015, 14, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Austin, P.J.; Moalem-Taylor, G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef]
- Calvo, M.; Dawes, J.M.; Bennett, D.L. The role of the immune system in the generation of neuropathic pain. Lancet Neurol. 2012, 11, 629–642. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Gensel, J.C.; Ankeny, D.P.; Alexander, J.K.; Donnelly, D.J.; Popovich, P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J. Neurosci. 2009, 29, 13435–13444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiguchi, N.; Kobayashi, Y.; Saika, F.; Sakaguchi, H.; Maeda, T.; Kishioka, S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain 2015, 156, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Pannell, M.; Labuz, D.; Celik, M.Ö.; Keye, J.; Batra, A.; Siegmund, B.; Machelska, H. Adoptive transfer of M2 macrophages reduces neuropathic pain via opioid peptides. J. Neuroinflamm. 2016, 13, 262. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.Y.; Gu, Y.; Chen, Y. Communication between neuronal somata and satellite glial cells in sensory ganglia. Glia 2013, 61, 1157–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, A.; Bennett, D.L. Neuroinflammation and the generation of neuropathic pain. Br. J. Anaesth. 2013, 111, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Richner, M.; Ulrichsen, M.; Elmegaard, S.L.; Dieu, R.; Pallesen, L.T.; Vaegter, C.B. Peripheral nerve injury modulates neurotrophin signaling in the peripheral and central nervous system. Mol. Neurobiol. 2014, 50, 945–970. [Google Scholar] [CrossRef]
- Tsujino, H.; Kondo, E.; Fukuoka, T.; Dai, Y.; Tokunaga, A.; Miki, K.; Yonenobu, K.; Ochi, T.; Noguchi, K. Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: A novel neuronal marker of nerve injury. Mol. Cell. Neurosci. 2000, 15, 170–182. [Google Scholar] [CrossRef]
- Averill, S.; Michael, G.J.; Shortland, P.J.; Leavesley, R.C.; King, V.R.; Bradbury, E.J.; McMahon, S.B.; Priestley, J.V. NGF and GDNF ameliorate the increase in ATF3 expression which occurs in dorsal root ganglion cells in response to peripheral nerve injury. Eur. J. Neurosci. 2004, 19, 1437–1445. [Google Scholar] [CrossRef]
- Mason, M.R.; Lieberman, A.R.; Anderson, P.N. Corticospinal neurons up-regulate a range of growth-associated genes following intracortical, but not spinal, axotomy. Eur. J. Neurosci. 2003, 18, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Nakagomi, S.; Suzuki, Y.; Namikawa, K.; Kiryu-Seo, S.; Kiyama, H. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J. Neurosci. 2003, 23, 5187–5196. [Google Scholar] [CrossRef] [PubMed]
- Deval, E.; Gasull, X.; Noël, J.; Salinas, M.; Baron, A.; Diochot, S.; Lingueglia, E. Acid-sensing ion channels (ASICs): Pharmacology and implication in pain. Pharm. Ther. 2010, 128, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Sun, W.H.; Chen, C.C. Genetic exploration of the role of acid-sensing ion channels. Neuropharmocology 2015, 94, 99–118. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, S.H.; Kim, Y.O.; Yoon, M.H. Antinociceptive effects of amiloride and benzamil in neuropathic pain model rats. J. Korean Med. Sci. 2013, 28, 1238–1243. [Google Scholar] [CrossRef]
- Kong, X.; Tang, X.; Du, W.; Tong, J.; Yan, Y.; Zheng, F.; Fang, M.; Gong, F.; Tan, Z. Extracellular acidosis modulates the endocytosis and maturation of macrophages. Cell Immunol. 2013, 281, 44–50. [Google Scholar] [CrossRef]
- Voilley, N.; de Weille, J.; Mamet, J.; Lazdunski, M. Nonsteroid Anti-Inflammatory Drugs inhibit both the activity and the inflammation-induced expression of acid-sensing ion channels in nociceptors. J. Neurosci. 2001, 21, 8026–8033. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Kolker, S.J.; Burnes, L.A.; Walder, R.Y.; Sluka, K.A. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain 2008, 137, 662–669. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.N.; Lee, C.H.; Lin, S.H.; Wong, C.W.; Sun, W.H.; Wood, J.N.; Chen, C.C. Roles of ASIC3, TRPV1, and NaV1.8 in the transition from acute to chronic pain in a mouse model of fibromyalgia. Mol. Pain. 2014, 10, 40. [Google Scholar] [CrossRef]
- Hsieh, W.S.; Kung, C.C.; Huang, S.L.; Lin, S.C.; Sun, W.H. TDAG8, TRPV1, and ASIC3 involved in establishing hyperalgesic priming in experimental rheumatoid arthritis. Sci. Rep. 2017, 7, 8870. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Chang, C.Y.; Chen, C.C.; Yang, C.D.; Sun, W.H. Distinct expression of Mas1-related G-protein-coupled receptor B4 in dorsal root and trigeminal ganglia-implications for altered behaviors in acid-sensing ion channel 3-deficient mice. J. Mol. Neurosci. 2013, 51, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Huang, C.W.; Lin, C.S.; Chang, W.H.; Sun, W.H. Expression and function of proton-sensing G protein-coupled receptors in inflammatory pain. Mol. Pain 2009, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.S.; Hartley, H.O. Biometrika Tables for Statisticians; Biometrika Trust: Oxford, UK, 1976; p. 289. [Google Scholar]
- Bennett, G.J.; Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 22, 87–107. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
- Gehrmann, J.; Monaco, S.; Kreutzberg, G.W. Spinal Cord microglial cells and DRG satellite cells rapidly respond to transection of the rat sciatic nerve. Restor. Neurol. Neurosci. 1991, 2, 181–198. [Google Scholar] [CrossRef]
- Liu, F.Y.; Sun, Y.N.; Wang, F.T.; Li, Q.; Su, L.; Zhao, Z.F.; Meng, X.L.; Zhao, H.; Wu, X.; Sun, Q.; et al. Activation of satellite glial cells in lumbar dorsal root ganglia contributes to neuropathic pain after spinal nerve ligation. Brain Res. 2012, 1427, 65–77. [Google Scholar] [CrossRef]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Seijffers, R.; Allchorne, A.J.; Woolf, C.J. The transcription factor ATF-3 promotes neurite outgrowth. Mol. Cell. Neurosci. 2006, 32, 143–154. [Google Scholar] [CrossRef]
- Kiryu-Seo, S.; Ohno, N.; Kidd, G.J.; Komuro, H.; Trapp, B.D. Demyelination increases axonal stationary mitochondrial size and the speed of axonal mitochrondrial transport. J. Neurosci. 2010, 306, 6586–6666. [Google Scholar]
- Hunt, D.; Hossain-Ibrahim, K.; Mason, M.R.; Coffin, R.S.; Lieberman, A.R.; Winterbottom, J.; Anderson, P.N. ATF3 upregulation in glia during Wallerian degeneration: Differential expression in peripheral nerves and CNS white matter. BMC Neurosci. 2004, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Mokarram, N.; Merchant, A.; Mukhatyar, V.; Patel, G.; Bellamkonda, R.V. Effect of Modulating Macrophage Phenotype on Peripheral Nerve Repair. Biomaterials 2012, 33, 8793–8801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, H.; Zhang, D.; Zhang, Y.; Wen, Y.; Wang, Y. ATF3 promotes migration and M1/M2 polarization of macrophages by activating tenascin-C via Wnt/β-catenin pathway. Mol. Med. Rep. 2017, 16, 3641–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basbaum, A.I.; Gautron, M.; Jazat, F.; Mayes, M.; Guilbaud, G. The spectrum of fiber loss in a model of neuropathic pain in the rat: An electron microscopic study. Pain 1991, 47, 359–367. [Google Scholar] [CrossRef]
- Ramer, M.S.; French, G.D.; Bisby, M.A. Wallerian degeneration is required for both neuropathic pain and sympathetic sprouting into the DRG. Pain 1997, 72, 71–78. [Google Scholar] [CrossRef]
- Schmid, A.B.; Coppieters, M.W.; Ruitenberg, M.J.; McLachlan, E.M. Local and Remote Immune-Mediated Inflammation After Mild Peripheral Nerve Compression in Rats. J. Neuropathol. Exp. Neurol. 2013, 72, 662–680. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.J.; Tandrup, T.; Bergman, E.; Xu, Z.Q.; Ulfhake, B.; Hökfelt, T. Effect of peripheral nerve injury on dorsal root ganglion neurons in the C57 BL/6J mouse: Marked changes both in cell numbers and neuropeptide expression. Neuroscience 2001, 105, 249–263. [Google Scholar] [CrossRef]
- Myers, R.R.; Heckman, H.M.; Rodriguez, M. Reduced Hyperalgesia in Nerve-Injured WLD Mice: Relationship to Nerve Fiber Phagocytosis, Axonal Degeneration, and Regeneration in Normal Mice. Exp. Neurol. 1996, 141, 94–101. [Google Scholar] [CrossRef]
- Sommer, C.; Schäfers, M. Painful Mononeuropathy in C57BL/Wld Mice with Delayed Wallerian Degeneration: Differential Effects of Cytokine Production and Nerve Regeneration on Thermal and Mechanical Hypersensitivity. Brain Res. 1998, 784, 154–162. [Google Scholar] [CrossRef]
- Coggeshall, R.E.; Dougherty, P.M.; Pover, C.M.; Carlton, S.M. Is large myelinated fiber loss associated with hyperalgesia in a model of experimental peripheral neuropathy in the rat? Pain 1993, 52, 233–242. [Google Scholar] [CrossRef]
- Brück, W. The role of macrophages in Wallerian degeneration. Brain Pathol. 1997, 7, 741–752. [Google Scholar] [CrossRef]
- Raggi, F.; Pelassa, S.; Pierobon, D.; Penco, F.; Gattorno, M.; Novelli, F.; Eva, A.; Varesio, L.; Giovarelli, M.; Bosco, M.C. Regulation of Human Macrophage M1-M2 Polarization Balance by Hypoxia and the Triggering Receptor Expressed on Myeloid Cells-1. Front. Immunol. 2017, 8, 1097. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Bae, D.J.; Kim, M.J.; Piao, M.L.; Kim, I.S. Extracellular low pH modulates phosphatidylserine-depedent phagocytosis in macrophages by increasing stabilin-1 expression. J. Biol. Chem. 2012, 287, 11261–11271. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kung, C.-C.; Huang, Y.-C.; Hung, T.-Y.; Teng, C.-Y.; Lee, T.-Y.; Sun, W.-H. Deletion of Acid-Sensing Ion Channel 3 Relieves the Late Phase of Neuropathic Pain by Preventing Neuron Degeneration and Promoting Neuron Repair. Cells 2020, 9, 2355. https://doi.org/10.3390/cells9112355
Kung C-C, Huang Y-C, Hung T-Y, Teng C-Y, Lee T-Y, Sun W-H. Deletion of Acid-Sensing Ion Channel 3 Relieves the Late Phase of Neuropathic Pain by Preventing Neuron Degeneration and Promoting Neuron Repair. Cells. 2020; 9(11):2355. https://doi.org/10.3390/cells9112355
Chicago/Turabian StyleKung, Chia-Chi, Yi-Chu Huang, Ting-Yun Hung, Chih-Yu Teng, Tai-Ying Lee, and Wei-Hsin Sun. 2020. "Deletion of Acid-Sensing Ion Channel 3 Relieves the Late Phase of Neuropathic Pain by Preventing Neuron Degeneration and Promoting Neuron Repair" Cells 9, no. 11: 2355. https://doi.org/10.3390/cells9112355
APA StyleKung, C. -C., Huang, Y. -C., Hung, T. -Y., Teng, C. -Y., Lee, T. -Y., & Sun, W. -H. (2020). Deletion of Acid-Sensing Ion Channel 3 Relieves the Late Phase of Neuropathic Pain by Preventing Neuron Degeneration and Promoting Neuron Repair. Cells, 9(11), 2355. https://doi.org/10.3390/cells9112355