Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches



Abstract
:1. Introduction
2. Introduction to Autoreactive B Cell Development and B Cell Subsets Described in CAD
2.1. Development of Autoreactive B Cells: Background Information on General Mechanisms
2.2. Overview of B Cell Subsets Described in CAD and Their Potential Role in Skin Autoimmunity
2.2.1. B-2 Cells
2.2.2. Memory B Cells
2.2.3. Plasma Cells (PCs)
2.2.4. Innate like B Cells (ILBs)
2.2.5. Regulatory B Cells (Bregs)
3. Potential Features and Clustering of B Cells in Lesional Skin
3.1. Role of B Cells as APC in Autoimmunity and Their Potential Contribution to Skin-Driven Inflammatory Responses
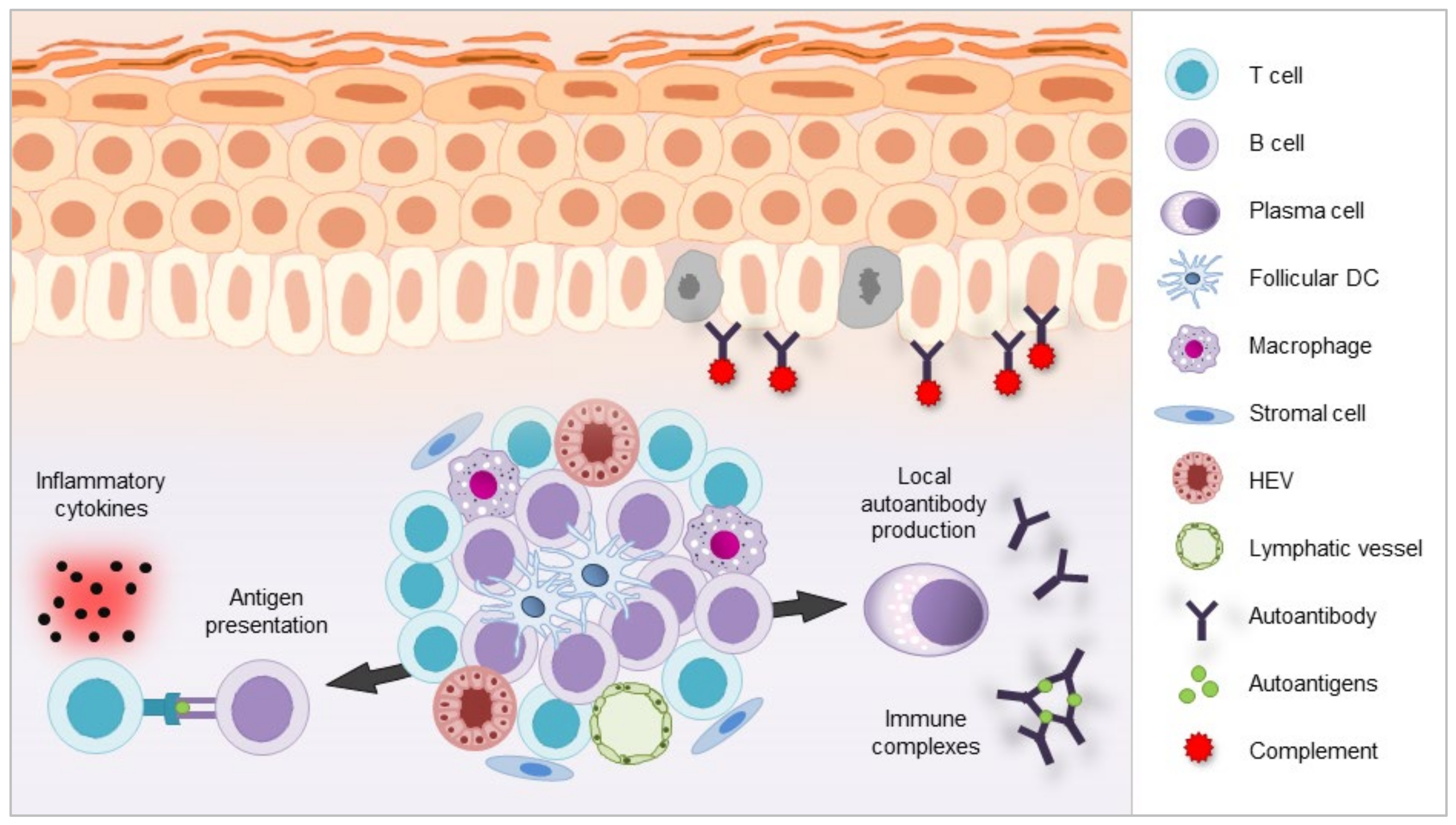
3.2. B Cells in the Formation of TLS
3.2.1. Historic Origins
3.2.2. Physiology of TLS
3.2.3. TLS Formation and Associated Inflammatory Cytokines
3.2.4. TLS Formation Provides a Niche for Local Autoimmunity
4. Traditional and Targeted Therapeutic Strategies in CAD
4.1. Traditional Therapeutic Approaches in CAD
4.2. B Cell Associated Therapeutic Strategies
4.2.1. Targeting B Cell Surface Molecules
CD 20 Antibodies: RTX
Second/Third Generation CD20 Antibodies
CD19 Antibodies
4.2.2. Targeting B Cell Activation and Survival Factors
BLyS (BAFF)/APRIL Antagonists
4.2.3. Targeting B Cell Signaling Molecules
BTK Inhibitors
PI3Kδ Inhibitors
SHIP1 Activators
ROCK2 Inhibitors
4.2.4. Challenges of Therapies Targeting B Cells
4.2.5. PC-Associated Therapeutic Strategies
Proteasome Inhibitors/Immunoproteasome Inhibitors
CXCR4 Antagonists
FcRn Receptor Antibodies
Plasmapheresis and Immunoabsorption
5. Summary and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brink, R. The imperfect control of self-reactive germinal center B cells. Curr. Opin. Immunol. 2014, 28, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Zenzo, G.; Di Lullo, G.; Corti, D.; Calabresi, V.; Sinistro, A.; Vanzetta, F.; Didona, B.; Cianchini, G.; Hertl, M.; Eming, R.; et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J. Clin. Investig. 2012, 122, 3781–3790. [Google Scholar] [CrossRef] [Green Version]
- DeFranco, A.L. Germinal centers and autoimmune disease in humans and mice. Immunol. Cell Biol. 2016, 94, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Winter, O.; Dame, C.; Jundt, F.; Hiepe, F. Pathogenic Long-Lived Plasma Cells and Their Survival Niches in Autoimmunity, Malignancy, and Allergy. J. Immunol. 2012, 189, 5105–5111. [Google Scholar] [CrossRef] [Green Version]
- Pan, M.; Liu, X.; Zheng, J. The pathogenic role of autoantibodies in pemphigus vulgaris. Clin. Exp. Dermatol. 2011, 36, 703–707. [Google Scholar] [CrossRef]
- Di Zenzo, G.; Della Torre, R.; Zambruno, G.; Borradori, L. Bullous pemphigoid: From the clinic to the bench. Clin. Dermatol. 2012, 30, 3–16. [Google Scholar] [CrossRef]
- Debes, G.F.; McGettigan, S.E. Skin-Associated B Cells in Health and Inflammation. J. Immunol. 2019, 202, 1659–1666. [Google Scholar] [CrossRef] [Green Version]
- Egbuniwe, I.U.; Karagiannis, S.N.; Nestle, F.O.; Lacy, K.E. Revisiting the role of B cells in skin immune surveillance. Trends Immunol. 2015, 36, 102–111. [Google Scholar] [CrossRef]
- Kabashima, K.; Honda, T.; Ginhoux, F.; Egawa, G. The immunological anatomy of the skin. Nat. Rev. Immunol. 2019, 19, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Corsiero, E.; Delvecchio, F.R.; Bombardieri, M.; Pitzalis, C. B cells in the formation of tertiary lymphoid organs in autoimmunity, transplantation and tumorigenesis. Curr. Opin. Immunol. 2019, 57, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Akashi, K.; Kondo, M.; Cheshier, S.; Shizuru, J.; Gandy, K.; Domen, J.; Mebius, R.; Traver, D.; Weissman, I. Lymphoid Development from Stem Cells and the Common Lymphocyte Progenitors. Cold Spring Harb. Symp. Quant. Biol. 1999, 64, 1–12. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Melchers, F. Checkpoints that control B cell development. J. Clin. Investig. 2015, 125, 2203–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Litinskiy, M.B.; Nardelli, B.; Hilbert, D.M.; He, B.; Schaffer, A.; Casali, P.; Cerutti, A. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat. Immunol. 2002, 3, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Jego, G.; Palucka, A.K.; Blanck, J.-P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid Dendritic Cells Induce Plasma Cell Differentiation through Type I Interferon and Interleukin. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Chan, T.D.; Wood, K.; Hermes, J.R.; Butt, D.; Jolly, C.J.; Basten, A.; Brink, R. Elimination of Germinal-Center-Derived Self-Reactive B Cells Is Governed by the Location and Concentration of Self-Antigen. Immunity 2012, 37, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.; Chung, Y.; Hwang, D.; Yang, X.O.; Kang, H.S.; Ma, L.; Wang, Y.-h.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Generation of T Follicular Helper Cells Is Mediated by Interleukin-21 but Independent of T Helper 1, 2, or 17 Cell Lineages. Immunity 2008, 29, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xu, H.; Shih, C.; Wan, Z.; Ma, X.; Ma, W.; Luo, D.; Qi, H. T–B-cell entanglement and ICOSL-driven feed-forward regulation of germinal centre reaction. Nat. Cell Biol. 2015, 517, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, A.; McGuire, H.; Yu, D.; Sprent, J.; Mackay, C.R.; King, C. A Fundamental Role for Interleukin-21 in the Generation of T Follicular Helper Cells. Immunity 2008, 29, 127–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zotos, D.; Coquet, J.M.; Zhang, Y.; Light, A.; D’Costa, K.; Kallies, A.; Corcoran, L.M.; Godfrey, D.I.; Toellner, K.-M.; Smyth, M.J.; et al. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell–intrinsic mechanism. J. Exp. Med. 2010, 207, 365–378. [Google Scholar] [CrossRef] [Green Version]
- Kawabe, T.; Naka, T.; Yoshida, K.; Tanaka, T.; Fujiwara, H.; Suematsu, S.; Yoshida, N.; Kishimoto, T.; Kikutani, H. The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1, 167–178. [Google Scholar] [CrossRef]
- Gitlin, A.D.; Shulman, Z.; Nussenzweig, M.C. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 2014, 509, 637–640. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Smith, D.; Aviszus, K.; Detanico, T.; Heiser, R.A.; Wysocki, L.J. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J. Exp. Med. 2010, 207, 2225–2237. [Google Scholar] [CrossRef] [Green Version]
- Shlomchik, M.; Mascelli, M.; Shan, H.; Radic, M.Z.; Pisetsky, D.; Marshak-Rothstein, A.; Weigert, M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 1990, 171, 265–292. [Google Scholar] [CrossRef] [Green Version]
- Kräutler, N.J.; Suan, D.; Butt, D.; Bourne, K.; Hermes, J.R.; Chan, T.D.; Sundling, C.; Kaplan, W.; Schofield, P.; Jackson, J.; et al. Differentiation of germinal center B cells into plasma cells is initiated by high-affinity antigen and completed by Tfh cells. J. Exp. Med. 2017, 214, 1259–1267. [Google Scholar] [CrossRef] [Green Version]
- Baumgarth, N. Innate-Like B Cells and Their Rules of Engagement. Adv. Exp. Med. Biol. 2013, 785, 57–66. [Google Scholar] [CrossRef]
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. [Google Scholar] [CrossRef]
- Inoue, T.; Moran, I.; Shinnakasu, R.; Phan, T.G.; Kurosaki, T. Generation of memory B cells and their reactivation. Immunol. Rev. 2018, 283, 138–149. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Matsushita, T.; Tedder, T.F. B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Ann. N. Y. Acad. Sci. 2010, 1183, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, K.A.; Kallies, A.; Nutt, S.L.; Tarlinton, D.M. Plasma cell development: From B-cell subsets to long-term survival niches. Semin. Immunol. 2008, 20, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Hussein, M.-R.A.; Aboulhagag, N.M.; Atta, H.S.; Atta, S.M. Evaluation of the profile of the immune cell infiltrate in lichen planus, discoid lupus erythematosus, and chronic dermatitis. Pathology 2008, 40, 682–693. [Google Scholar] [CrossRef]
- O’Brien, J.C.; Hosler, G.A.; Chong, B.F. Changes in T cell and B cell composition in discoid lupus erythematosus skin at different stages. J. Dermatol. Sci. 2017, 85, 247–249. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Zhou, S.; Liu, Z.; Cong, W.; Fei, X.; Zeng, W.; Zhu, H.; Xu, R.; Wang, Y.; Zheng, J.; et al. Pivotal Role of Lesional and Perilesional T/B Lymphocytes in Pemphigus Pathogenesis. J. Investig. Dermatol. 2017, 137, 2362–2370. [Google Scholar] [CrossRef] [Green Version]
- Bosello, S.; Angelucci, C.; Lama, G.; Alivernini, S.; Proietti, G.; Tolusso, B.; Sica, G.; Gremese, E.; Ferraccioli, G. Characterization of inflammatory cell infiltrate of scleroderma skin: B cells and skin score progression. Arthritis Res. Ther. 2018, 20, 1–11. [Google Scholar] [CrossRef]
- Lu, J.; Ding, Y.; Yi, X.; Zheng, J. CD19+ B cell subsets in the peripheral blood and skin lesions of psoriasis patients and their correlations with disease severity. Braz. J. Med Biol. Res. 2016, 49, e5374. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, F.; Abul, H.; Al Saleh, Q.; Hassab-el Naby, H.; Kajeji, M.; Haines, D.; Burleson, J.; Morgan, G. Elevated B-Lymphocyte Levels in Lesional Tissue of Non-Arthritic Psoriasis. J. Dermatol. 1999, 26, 428–433. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Coussens, L.M. B cells and their mediators as targets for therapy in solid tumors. Exp. Cell Res. 2013, 319, 1644–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Mei, H.; Dörner, T.; Hiepe, F.; Radbruch, A.; Fillatreau, S.; Hoyer, B.F. Memory B and memory plasma cells. Immunol. Rev. 2010, 237, 117–139. [Google Scholar] [CrossRef]
- Ochsenbein, A.F.; Pinschewer, D.D.; Sierro, S.; Horvath, E.; Hengartner, H.; Zinkernagel, R.M. Protective long-term antibody memory by antigen-driven and T help-dependent differentiation of long-lived memory B cells to short-lived plasma cells independent of secondary lymphoid organs. Proc. Natl. Acad. Sci. USA 2000, 97, 13263–13268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, K.A.; Taylor, J.J.; Maul, R.W.; Gearhart, P.J.; Jenkins, M.K. Different B Cell Populations Mediate Early and Late Memory During an Endogenous Immune Response. Science 2011, 331, 1203–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct Effector Cytokine Profiles of Memory and Naive Human B Cell Subsets and Implication in Multiple Sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harp, C.T.; Ireland, S.; Davis, L.S.; Remington, G.; Cassidy, B.; Cravens, P.D.; Stuve, O.; Lovett-Racke, A.E.; Eagar, T.N.; Greenberg, B.M.; et al. Memory B cells from a subset of treatment-naïve relapsing-remitting multiple sclerosis patients elicit CD4+ T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 2010, 40, 2942–2956. [Google Scholar] [CrossRef]
- Benson, M.J.; Dillon, S.R.; Castigli, E.; Geha, R.S.; Xu, S.; Lam, K.-P.; Noelle, R.J. Cutting Edge: The Dependence of Plasma Cells and Independence of Memory B Cells on BAFF and APRIL. J. Immunol. 2008, 180, 3655–3659. [Google Scholar] [CrossRef] [Green Version]
- Stohl, W.; Hiepe, F.; Latinis, K.M.; Thomas, M.; Scheinberg, M.A.; Clarke, A.; Aranow, C.; Wellborne, F.R.; Abud-Mendoza, C.; Hough, D.R.; et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Pollmann, R.; Walter, E.; Schmidt, T.; Waschke, J.; Hertl, M.; Möbs, C.; Eming, R. Identification of Autoreactive B Cell Subpopulations in Peripheral Blood of Autoimmune Patients With Pemphigus Vulgaris. Front. Immunol. 2019, 10, 1375. [Google Scholar] [CrossRef] [Green Version]
- Nishifuji, K.; Amagai, M.; Kuwana, M.; Iwasaki, T.; Nishikawa, T. Detection of Antigen-Specific B Cells in Patients with Pemphigus Vulgaris by Enzyme-Linked Immunospot Assay: Requirement of T Cell Collaboration for Autoantibody Production. J. Investig. Dermatol. 2000, 114, 88–94. [Google Scholar] [CrossRef]
- Hennerici, T.; Pollmann, R.; Schmidt, T.; Seipelt, M.; Tackenberg, B.; Möbs, C.; Ghoreschi, K.; Hertl, M.; Eming, R. Increased Frequency of T Follicular Helper Cells and Elevated Interleukin-27 Plasma Levels in Patients with Pemphigus. PLoS ONE 2016, 11, e0148919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paus, D.; Phan, T.G.; Chan, T.D.; Gardam, S.; Basten, A.; Brink, R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006, 203, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manz, R.A.; Thiel, A.; Radbruch, A. Lifetime of plasma cells in the bone marrow. Nat. Cell Biol. 1997, 388, 133–134. [Google Scholar] [CrossRef]
- Manz, R.A.; Löhning, M.; Cassese, G.; Thiel, A.; Radbruch, A. Survival of long-lived plasma cells is independent of antigen [In Process Citation]. Int. Immunol. 1998, 10, 1703–1711. [Google Scholar] [CrossRef]
- Ahuja, A.; Anderson, S.M.; Khalil, A.; Shlomchik, M.J. Maintenance of the plasma cell pool is independent of memory B cells. Proc. Natl. Acad. Sci. USA 2008, 105, 4802–4807. [Google Scholar] [CrossRef] [Green Version]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral Immunity Due to Long-Lived Plasma Cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Cassese, G.; Arce, S.; Hauser, A.E.; Lehnert, K.; Moewes, B.; Mostarac, M.; Muehlinghaus, G.; Szyska, M.; Radbruch, A.; Manz, R.A. Plasma Cell Survival Is Mediated by Synergistic Effects of Cytokines and Adhesion-Dependent Signals. J. Immunol. 2003, 171, 1684–1690. [Google Scholar] [CrossRef]
- Matthes, T.; Dunand-Sauthier, I.; Santiago-Raber, M.-L.; Krause, K.-H.; Donze, O.; Passweg, J.; McKee, T.; Huard, B. Production of the plasma-cell survival factor a proliferation-inducing ligand (APRIL) peaks in myeloid precursor cells from human bone marrow. Blood 2011, 118, 1838–1844. [Google Scholar] [CrossRef]
- Thai, L.-H.; Le Gallou, S.; Robbins, A.; Crickx, E.; Fadeev, T.; Zhou, Z.; Cagnard, N.; Mégret, J.; Bole, C.; Weill, J.-C.; et al. BAFF and CD4+ T cells are major survival factors for long-lived splenic plasma cells in a B-cell–depletion context. Blood 2018, 131, 1545–1555. [Google Scholar] [CrossRef]
- Dong, W.; Li, X.; Liu, H.; Zhu, P. Infiltrations of plasma cells in synovium are highly associated with synovial fluid levels of APRIL in inflamed peripheral joints of rheumatoid arthritis. Rheumatol. Int. 2009, 29, 801–806. [Google Scholar] [CrossRef]
- Tiburzy, B.; Szyska, M.; Iwata, H.; Chrobok, N.; Kulkarni, U.; Hirose, M.; Ludwig, R.J.; Kalies, K.; Westermann, J.; Wong, D.; et al. Persistent Autoantibody-Production by Intermediates between Short-and Long-Lived Plasma Cells in Inflamed Lymph Nodes of Experimental Epidermolysis Bullosa Acquisita. PLoS ONE 2013, 8, e83631. [Google Scholar] [CrossRef] [PubMed]
- Geherin, S.A.; Fintushel, S.R.; Lee, M.H.; Wilson, R.P.; Patel, R.T.; Alt, C.; Young, A.J.; Hay, J.B.; Debes, G.F. The Skin, a Novel Niche for Recirculating B Cells. J. Immunol. 2012, 188, 6027–6035. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Konishi, H.; Ito, M.; Nagura, H.; Asai, J. Identification of Secretory Immunoglobulin A in Human Sweat and Sweat Glands. J. Investig. Dermatol. 1988, 90, 648–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metze, D.; Jurecka, W.; Gebhart, W.; Schuller-Petrovic, S. Secretory immunoglobulin A in sweat gland tumors. J. Cutan. Pathol. 1989, 16, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Karaaslan, S.; Tomayko, M.M. A Niche for Plasma Cells: The Skin. J. Investig. Dermatol. 2019, 139, 2411–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.P.; McGettigan, S.E.; van Dang, D.; Kumar, A.; Cancro, M.P.; Nikbakht, N.; Stohl, W.; Debes, G.F. IgM Plasma Cells Reside in Healthy Skin and Accumulate with Chronic Inflammation. J. Investig. Dermatol. 2019, 139, 2477–2487. [Google Scholar] [CrossRef]
- Wenzel, J.; Landmann, A.; Vorwerk, G.; Kuhn, A. High expression of B lymphocyte stimulator in lesional keratinocytes of patients with cutaneous lupus erythematosus. Exp. Dermatol. 2018, 27, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, M.; Long, D.; Li, Q.; Zhao, M.; Wu, H.; Lu, Q. Abnormal expression of BAFF and its receptors in peripheral blood and skin lesions from systemic lupus erythematosus patients. Autoimmun 2020, 53, 192–200. [Google Scholar] [CrossRef]
- Aziz, M.; Holodick, N.E.; Rothstein, T.L.; Wang, P. The role of B-1 cells in inflammation. Immunol. Res. 2015, 63, 153–166. [Google Scholar] [CrossRef]
- Rothstein, T.L.; Quach, T.D. The human counterpart of mouse B-1 cells. Ann. N. Y. Acad. Sci. 2015, 1362, 143–152. [Google Scholar] [CrossRef]
- Montecino-Rodriguez, E.; Leathers, H.; Dorshkind, K. Identification of a B-1 B cell–specified progenitor. Nat. Immunol. 2006, 7, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Tsay, G.J.; Zouali, M. The Interplay between Innate-Like B Cells and Other Cell Types in Autoimmunity. Front. Immunol. 2018, 9, 1064. [Google Scholar] [CrossRef] [Green Version]
- Kaveri, S.V.; Silverman, G.J.; Bayry, J. Natural IgM in Immune Equilibrium and Harnessing Their Therapeutic Potential. J. Immunol. 2012, 188, 939–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boes, M.; Schmidt, T.; Linkemann, K.; Beaudette, B.C.; Marshak-Rothstein, A.; Chen, J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. USA 2000, 97, 1184–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, B.; Croker, B.P.; Morel, L. Lupus resistance is associated with marginal zone abnormalities in an NZM murine model. Lab. Investig. 2007, 87, 14–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, B.; Morel, L. Role of B-1a cells in autoimmunity. Autoimmun. Rev. 2006, 5, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.O.; Rothstein, T.L. A small CD11b+ human B1 cell subpopulation stimulates T cells and is expanded in lupus. J. Exp. Med. 2011, 208, 2591–2598. [Google Scholar] [CrossRef] [Green Version]
- Kessel, A.; Haj, T.; Peri, R.; Snir, A.; Melamed, D.; Sabo, E.; Toubi, E. Human CD19+CD25high B regulatory cells suppress proliferation of CD4+ T cells and enhance Foxp3 and CTLA-4 expression in T-regulatory cells. Autoimmun. Rev. 2012, 11, 670–677. [Google Scholar] [CrossRef]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; van Dang, D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nat. Cell Biol. 2014, 507, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Fillatreau, S.; Gray, D.; Anderton, S.M. Not always the bad guys: B cells as regulators of autoimmune pathology. Nat. Rev. Immunol. 2008, 8, 391–397. [Google Scholar] [CrossRef]
- Hilgenberg, E.; Shen, P.; Dang, V.D.; Ries, S.; Sakwa, I.; Fillatreau, S. Interleukin-10-Producing B Cells and the Regulation of Immunity. Curr. Top. Microbiol. Immunol. 2014, 380, 69–92. [Google Scholar] [CrossRef]
- Fillatreau, S. Novel regulatory functions for Toll-like receptor-activated B cells during intracellular bacterial infection. Immunol. Rev. 2011, 240, 52–71. [Google Scholar] [CrossRef]
- Fillatreau, S. Regulatory plasma cells. Curr. Opin. Pharmacol. 2015, 23, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Regulatory functions of innate-like B cells. Cell. Mol. Immunol. 2013, 10, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauri, C.; Menon, M. The expanding family of regulatory B cells. Int. Immunol. 2015, 27, 479–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Baba, A.; Yokota, T.; Nishikawa, H.; Ohkawa, Y.; Kayama, H.; Kallies, A.; Nutt, S.L.; Sakaguchi, S.; Takeda, K.; et al. Interleukin-10-Producing Plasmablasts Exert Regulatory Function in Autoimmune Inflammation. Immunity 2014, 41, 1040–1051. [Google Scholar] [CrossRef] [Green Version]
- Katz, S.I.; Parker, D.; Turk, J.L. B-cell suppression of delayed hypersensitivity reactions. Nature 1974, 251, 550–551. [Google Scholar] [CrossRef]
- Neta, R.; Salvin, S.B. Specific suppression of delayed hypersensitivity: The possible presence of a suppressor B cell in the regulation of delayed hypersensitivity. J. Immunol. 1974, 113, 1716–1725. [Google Scholar]
- Geherin, S.A.; Gómez, D.; Glabman, R.A.; Ruthel, G.; Hamann, A.; Debes, G.F. IL-10+Innate-like B Cells Are Part of the Skin Immune System and Require α4β1 Integrin To Migrate between the Peritoneum and Inflamed Skin. J. Immunol. 2016, 196, 2514–2525. [Google Scholar] [CrossRef] [Green Version]
- Dass, S.; Vital, E.M.; Emery, P. Development of psoriasis after B cell depletion with rituximab. Arthritis Rheum. 2007, 56, 2715–2718. [Google Scholar] [CrossRef]
- Fiorillo, L.; Wang, C.; Hemmati, I. Rituximab Induced Psoriasis in an Infant. Pediatr. Dermatol. 2014, 31, e149–e151. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Canoui-Poitrine, F.; Gottenberg, J.-E.; Economu-Dubosc, A.; Medkour, F.; Chevalier, X.; Bastuji-Garin, S.; Le Louet, H.; Farrenq, V.; Claudepierre, P. Incidence of New-onset and Flare of Preexisting Psoriasis During Rituximab Therapy for Rheumatoid Arthritis: Data from the French AIR Registry. J. Rheumatol. 2012, 39, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Guidelli, G.M.; Fioravanti, A.; Rubegni, P.; Feci, L. Induced psoriasis after rituximab therapy for rheumatoid arthritis: A case report and review of the literature. Rheumatol. Int. 2012, 33, 2927–2930. [Google Scholar] [CrossRef] [PubMed]
- Alahmari, H.S.; Alhowaish, N.Y.; Omair, M.A. Rituximab-Induced Psoriasis in a Patient with Granulomatosis with Polyangitis Treated with Adalimumab. Case Rep. Rheumatol. 2019, 2019. [Google Scholar] [CrossRef]
- Kim, D.-W.; Park, S.-K.; Woo, S.-H.; Yun, S.-K.; Kim, H.-U.; Park, J. New-onset psoriasis induced by rituximab therapy for non-Hodgkin lymphoma in a child. Eur. J. Dermatol. 2016, 26, 190–191. [Google Scholar] [CrossRef]
- Li, W.; Tian, X.; Lu, X.; Peng, Q.; Shu, X.; Yang, H.; Li, Y.; Wang, Y.; Zhang, X.; Liu, Q.; et al. Significant decrease in peripheral regulatory B cells is an immunopathogenic feature of dermatomyositis. Sci. Rep. 2016, 6, 27479. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.-Q.; Xu, R.-C.; Chen, Y.-Y.; Yuan, H.-J.; Cao, H.; Zhao, X.-Q.; Zheng, J.; Wang, Y.; Pan, M. Impaired function of CD19+CD24hiCD38hiregulatory B cells in patients with pemphigus. Br. J. Dermatol. 2015, 172, 101–110. [Google Scholar] [CrossRef]
- Crawford, A.; MacLeod, M.; Schumacher, T.; Corlett, L.; Gray, D. Primary T Cell Expansion and Differentiation In Vivo Requires Antigen Presentation by B Cells. J. Immunol. 2006, 176, 3498–3506. [Google Scholar] [CrossRef] [Green Version]
- Linton, P.-J.; Harbertson, J.; Bradley, L.M. A Critical Role for B Cells in the Development of Memory CD4 Cells. J. Immunol. 2000, 165, 5558–5565. [Google Scholar] [CrossRef]
- Clatza, A.; Bonifaz, L.C.; Vignali, D.A.A.; Moreno, J. CD40-Induced Aggregation of MHC Class II and CD80 on the Cell Surface Leads to an Early Enhancement in Antigen Presentation. J. Immunol. 2003, 171, 6478–6487. [Google Scholar] [CrossRef]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, Z.; Rozo, C.T.; Hamed, H.A.; Alem, F.; Urban, J.F.; Gause, W.C. The Role of B Cells in the Development of CD4 Effector T Cells during a Polarized Th2 Immune Response. J. Immunol. 2007, 179, 3821–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, S.K.; Cao, Y.; Hamel, K.M.; Doodes, P.D.; Hutas, G.; Finnegan, A. Expression of CD80/86 on B cells is essential for autoreactive T cell activation and the development of arthritis. J. Immunol. 2007, 179, 5109–5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of Effector CD4 T Cell Populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [Green Version]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A Novel Mouse with B Cells but Lacking Serum Antibody Reveals an Antibody-independent Role for B Cells in Murine Lupus. J. Exp. Med. 1999, 189, 1639–1648. [Google Scholar] [CrossRef]
- Giles, J.R.; Kashgarian, M.; Koni, P.A.; Shlomchik, M.J. B Cell-Specific MHC Class II Deletion Reveals Multiple Nonredundant Roles for B Cell Antigen Presentation in Murine Lupus. J. Immunol. 2015, 195, 2571–2579. [Google Scholar] [CrossRef] [Green Version]
- Eming, R.; Nagel, A.; Wolff-Franke, S.; Podstawa, E.; Debus, D.; Hertl, M. Rituximab Exerts a Dual Effect in Pemphigus Vulgaris. J. Investig. Dermatol. 2008, 128, 2850–2858. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-P.; Wu, J.; Han, Y.-F.; Shi, Z.-R.; Wang, L. Pathogenesis of cutaneous lupus erythema associated with and without systemic lupus erythema. Autoimmun. Rev. 2017, 16, 735–742. [Google Scholar] [CrossRef]
- Wenzel, J. Cutaneous lupus erythematosus: New insights into pathogenesis and therapeutic strategies. Nat. Rev. Rheumatol. 2019, 15, 519–532. [Google Scholar] [CrossRef]
- Kogame, T.; Yamashita, R.; Hirata, M.; Kataoka, T.R.; Kamido, H.; Ueshima, C.; Matsui, M.; Nomura, T.; Kabashima, K. Analysis of possible structures of inducible skin-associated lymphoid tissue in lupus erythematosus profundus. J. Dermatol. 2018, 45, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Liu, Z.; Yuan, H.; Zhao, X.; Zou, Y.; Zheng, J.; Pan, M. Autoreactive B Cell Differentiation in Diffuse Ectopic Lymphoid-Like Structures of Inflamed Pemphigus Lesions. J. Investig. Dermatol. 2020, 140, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Streilein, J.W. Skin-Associated Lymphoid Tissues (SALT): Origins and Functions. J. Investig. Dermatol. 1983, 80, 12s–16s. [Google Scholar] [CrossRef] [PubMed]
- Streilein, J.W. Circuits and Signals of the Skin-Associated Lymphoid Tissues (SALT). J. Investig. Dermatol. 1985, 85, S10–S13. [Google Scholar] [CrossRef] [Green Version]
- Natsuaki, Y.; Egawa, G.; Nakamizo, S.; Ono, S.; Hanakawa, S.; Okada, T.; Kusuba, N.; Otsuka, A.; Kitoh, A.; Honda, T.; et al. Perivascular leukocyte clusters are essential for efficient activation of effector T cells in the skin. Nat. Immunol. 2014, 15, 1064–1069. [Google Scholar] [CrossRef]
- Barone, F.; Gardner, D.H.; Nayar, S.; Steinthal, N.; Buckley, C.D.; Luther, S.A. Stromal Fibroblasts in Tertiary Lymphoid Structures: A Novel Target in Chronic Inflammation. Front. Immunol. 2016, 7, 477. [Google Scholar] [CrossRef]
- Ruddle, N.H. High Endothelial Venules and Lymphatic Vessels in Tertiary Lymphoid Organs: Characteristics, Functions, and Regulation. Front. Immunol. 2016, 7, 491. [Google Scholar] [CrossRef] [Green Version]
- Ruddle, N.H. Lymphatic vessels and tertiary lymphoid organs. J. Clin. Investig. 2014, 124, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Stranford, S.; Ruddle, N.H. Follicular dendritic cells, conduits, lymphatic vessels, and high endothelial venules in tertiary lymphoid organs: Parallels with lymph node stroma. Front. Immunol. 2012, 3, 350. [Google Scholar] [CrossRef] [Green Version]
- Link, A.; Hardie, D.L.; Favre, S.; Britschgi, M.R.; Adams, D.H.; Sixt, M.; Cyster, J.G.; Buckley, C.D.; Luther, S.A. Association of T-Zone Reticular Networks and Conduits with Ectopic Lymphoid Tissues in Mice and Humans. Am. J. Pathol. 2011, 178, 1662–1675. [Google Scholar] [CrossRef] [Green Version]
- Drayton, D.L.; Ying, X.; Lee, J.; Lesslauer, W.; Ruddle, N.H. Ectopic LTαβ Directs Lymphoid Organ Neogenesis with Concomitant Expression of Peripheral Node Addressin and a HEV-restricted Sulfotransferase. J. Exp. Med. 2003, 197, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Kratz, A.; Campos-Neto, A.; Hanson, M.S.; Ruddle, N.H. Chronic inflammation caused by lymphotoxin is lymphoid neogenesis. J. Exp. Med. 1996, 183, 1461–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmsen, A.; Kusser, K.; Hartson, L.; Tighe, M.; Sunshine, M.J.; Sedgwick, J.D.; Choi, Y.; Littman, D.R.; Randall, T.D. Cutting Edge: Organogenesis of Nasal-Associated Lymphoid Tissue (NALT) Occurs Independently of Lymphotoxin-α (LTα) and Retinoic Acid Receptor-Related Orphan Receptor-γ, but the Organization of NALT Is LTα Dependent. J. Immunol. 2002, 168, 986–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pipi, E.; Nayar, S.; Gardner, D.H.; Colafrancesco, S.; Smith, C.; Barone, F. Tertiary Lymphoid Structures: Autoimmunity Goes Local. Front. Immunol. 2018, 9, 1952. [Google Scholar] [CrossRef] [PubMed]
- Szyszko, E.A.; Brokstad, K.A.; Oijordsbakken, G.; Jonsson, M.V.; Jonsson, R.; Skarstein, K. Salivary glands of primary Sjogren’s syndrome patients express factors vital for plasma cell survival. Arthritis Res. Ther. 2011, 13, R2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, F.; Nayar, S.; Campos, J.; Cloake, T.; Withers, D.R.; Toellner, K.-M.; Zhang, Y.; Fouser, L.A.; Fisher, B.A.; Bowman, S.; et al. IL-22 regulates lymphoid chemokine production and assembly of tertiary lymphoid organs. Proc. Natl. Acad. Sci. USA 2015, 112, 11024–11029. [Google Scholar] [CrossRef] [Green Version]
- Rangel-Moreno, J.; Carragher, D.M.; La Luz Garcia-Hernandez, M.d.; Hwang, J.Y.; Kusser, K.; Hartson, L.; Kolls, J.K.; Khader, S.A.; Randall, T.D. The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat. Immunol. 2011, 12, 639–646. [Google Scholar] [CrossRef]
- Pitzalis, C.; Jones, G.W.; Bombardieri, M.; Jones, S.A. Ectopic lymphoid-like structures in infection, cancer and autoimmunity. Nat. Rev. Immunol. 2014, 14, 447–462. [Google Scholar] [CrossRef]
- Buckley, C.D.; Barone, F.; Nayar, S.; Bénézech, C.; Caamaño, J. Stromal Cells in Chronic Inflammation and Tertiary Lymphoid Organ Formation. Annu. Rev. Immunol. 2015, 33, 715–745. [Google Scholar] [CrossRef]
- Vos, K.; Thurlings, R.M.; Wijbrandts, C.A.; Van Schaardenburg, D.; Gerlag, D.M.; Tak, P.P. Early effects of rituximab on the synovial cell infiltrate in patients with rheumatoid arthritis. Arthritis Rheum. 2007, 56, 772–778. [Google Scholar] [CrossRef]
- Quartuccio, L.; Fabris, M.; Moretti, M.; Barone, F.; Bombardieri, M.; Rupolo, M.; Lombardi, S.; Pitzalis, C.; Beltrami, C.A.; Curcio, F.; et al. Resistance to Rituximab Therapy and Local BAFF Overexpression in Sjögren’s Syndrome-Related Myoepithelial Sialadenitis and Low-Grade Parotid B-Cell Lymphoma. Open Rheumatol. J. 2008, 2, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, K.; Clauder, A.-K.; Manz, R.A. Targeting B Cells and Plasma Cells in Autoimmune Diseases. Front. Immunol. 2018, 9, 835. [Google Scholar] [CrossRef] [PubMed]
- Massone, C.; Kodama, K.; Salmhofer, W.; Abe, R.; Shimizu, H.; Parodi, A.; Kerl, H.; Cerroni, L. Lupus erythematosus panniculitis (lupus profundus): Clinical, histopathological, and molecular analysis of nine cases. J. Cutan. Pathol. 2005, 32, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Egami, S.; Yamagami, J.; Amagai, M. Autoimmune bullous skin diseases, pemphigus and pemphigoid. J. Allergy Clin. Immunol. 2020, 145, 1031–1047. [Google Scholar] [CrossRef]
- Liu, Z.; Diaz, L.A.; Troy, J.L.; Taylor, A.F.; Emery, D.J.; Fairley, J.A.; Giudice, G.J. A passive transfer model of the organ-specific autoimmune disease, bullous pemphigoid, using antibodies generated against the hemidesmosomal antigen, BP180. J. Clin. Investig. 1993, 92, 2480–2488. [Google Scholar] [CrossRef]
- Amagai, M.; Kárpáti, S.; Prussick, R.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J. Clin. Investig. 1992, 90, 919–926. [Google Scholar] [CrossRef]
- Thoma-Uszynski, S.; Uter, W.; Schwietzke, S.; Schuler, G.; Borradori, L.; Hertl, M. Autoreactive T and B Cells from Bullous Pemphigoid (BP) Patients Recognize Epitopes Clustered in Distinct Regions of BP180 and BP180 and BP230. J. Immunol. 2006, 176, 2015–2023. [Google Scholar] [CrossRef] [Green Version]
- Penha, M.Á.; Farat, J.G.; Miot, H.A.; Barraviera, S.R.C.S. Quality of life index in autoimmune bullous dermatosis patients. An. Bras. Dermatol. 2015, 90, 190–194. [Google Scholar] [CrossRef]
- Jain, S.V.; Murrell, D.F. Psychosocial impact of inherited and autoimmune blistering diseases. Int. J. Women’s Dermatol. 2018, 4, 49–53. [Google Scholar] [CrossRef]
- Wang, E.Q.; Radjenovic, M.; Castrillon, M.A.; Feng, G.; Murrell, D.F. The effect of autoimmune blistering diseases on work productivity. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 1959–1966. [Google Scholar] [CrossRef]
- Kuhn, A.; Herrmann, M.; Kleber, S.; Beckmann-Welle, M.; Fehsel, K.; Martin-Villalba, A.; Lehmann, P.; Ruzicka, T.; Krammer, P.H.; Kolb-Bachofen, V. Accumulation of apoptotic cells in the epidermis of patients with cutaneous lupus erythematosus after ultraviolet irradiation. Arthritis Rheum. 2006, 54, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Scholtissek, B.; Zahn, S.; Maier, J.; Klaeschen, S.; Braegelmann, C.; Hoelzel, M.; Bieber, T.; Barchet, W.; Wenzel, J. Immunostimulatory Endogenous Nucleic Acids Drive the Lesional Inflammation in Cutaneous Lupus Erythematosus. J. Investig. Dermatol. 2017, 137, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A.; Hsu, H.-C.; Mountz, J.D. Autoreactive B cells in SLE, villains or innocent bystanders? Immunol. Rev. 2019, 292, 120–138. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.C.; Nesbitt, L.T. UPDATE ON SYSTEMIC GLUCOCORTICOSTEROIDS IN DERMATOLOGY. Dermatol. Clin. 2001, 19, 63–77. [Google Scholar] [CrossRef]
- Brattsand, R.; Linden, M. Cytokine modulation by glucocorticoids: Mechanisms and actions in cellular studies. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. S2), 81–90, discussion 91–92. [Google Scholar] [CrossRef]
- Olnes, M.J.; Kotliarov, Y.; Biancotto, A.; Cheung, F.; Chen, J.; Shi, R.; Zhou, H.; Wang, E.; Tsang, J.S.; Nussenblatt, R. Effects of Systemically Administered Hydrocortisone on the Human Immunome. Sci. Rep. 2016, 6, 23002. [Google Scholar] [CrossRef] [Green Version]
- Didona, D.; Maglie, R.; Eming, R.; Hertl, M. Pemphigus: Current and Future Therapeutic Strategies. Front. Immunol. 2019, 10, 1418. [Google Scholar] [CrossRef] [Green Version]
- Chams-Davatchi, C.; Mortazavizadeh, A.; Daneshpazhooh, M.; Davatchi, F.; Balighi, K.; Esmaili, N.; Akhyani, M.; Hallaji, Z.; Seirafi, H.; Mortazavi, H. Randomized double blind trial of prednisolone and azathioprine, vs. prednisolone and placebo, in the treatment of pemphigus vulgaris. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 1285–1292. [Google Scholar] [CrossRef]
- Ratnam, K.V.; Phay, K.L.; Tan, C.K. Pemphigus Therapy with Oral Prednisolone Regimens A 5-Year Study. Int. J. Dermatol. 1990, 29, 363–367. [Google Scholar] [CrossRef]
- Buchman, A.L. Side Effects of Corticosteroid Therapy. J. Clin. Gastroenterol. 2001, 33, 289–294. [Google Scholar] [CrossRef]
- Hoyer, B.F.; Moser, K.; Hauser, A.E.; Peddinghaus, A.; Voigt, C.; Eilat, D.; Radbruch, A.; Hiepe, F.; Manz, R.A. Short-lived Plasmablasts and Long-lived Plasma Cells Contribute to Chronic Humoral Autoimmunity in NZB/W Mice. J. Exp. Med. 2004, 199, 1577–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; O’Brien, T.; Yap, L.M.; Prince, H.M.; McCormack, C.J. The use of methotrexate in dermatology: A review. Australas. J. Dermatol. 2011, 53, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chan, J.J. Cyclophosphamide in dermatology. Australas. J. Dermatol. 2017, 58, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Eugui, E.M. Immunosuppressive and other Effects of Mycophenolic Acid and an Ester Prodrug, Mycophenolate Mofetil. Immunol. Rev. 1993, 136, 5–28. [Google Scholar] [CrossRef]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schlüter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Eickenberg, S.; Mickholz, E.; Jung, E.; Nofer, J.-R.; Pavenstädt, H.; Jacobi, A.M. Mycophenolic acid counteracts B cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R110. [Google Scholar] [CrossRef] [Green Version]
- Beissert, S.; Werfel, T.; Frieling, U.; Böhm, M.; Sticherling, M.; Stadler, R.; Zillikens, D.; Rzany, B.; Hunzelmann, N.; Meurer, M.; et al. A Comparison of Oral Methylprednisolone Plus Azathioprine or Mycophenolate Mofetil for the Treatment of Pemphigus. Arch. Dermatol. 2006, 142, 1447–1454. [Google Scholar] [CrossRef] [Green Version]
- Allison, A.C. Mechanisms of action of mycophenolate mofetil. Lupus 2005, 14 (Suppl. S1), s2–s8. [Google Scholar] [CrossRef]
- Guenther, L.; Lynde, C.; Poulin, Y. Off-Label Use of Topical Calcineurin Inhibitors in Dermatologic Disorders. J. Cutan. Med. Surg. 2019, 23, 27S–34S. [Google Scholar] [CrossRef]
- Hammer, O. CD19 as an attractive target for antibody-based therapy. mAbs 2012, 4, 571–577. [Google Scholar] [CrossRef] [Green Version]
- Du, F.H.; Mills, E.A.; Mao-Draayer, Y. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Autoimmun. Highlights 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Hilbert, D.M. The discovery and development of belimumab: The anti-BLyS–lupus connection. Nat. Biotechnol. 2012, 30, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Einhaus, J.; Pecher, A.-C.; Asteriti, E.; Schmid, H.; Secker, K.-A.; Duerr-Stoerzer, S.; Keppeler, H.; Klein, R.; Schneidawind, C.; Henes, J.; et al. Inhibition of effector B cells by ibrutinib in systemic sclerosis. Arthritis Res. 2020, 22, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, K.D.; Gold, M.R. Selective inhibitors of phosphoinositide 3-kinase delta: Modulators of B-cell function with potential for treating autoimmune inflammatory diseases and B-cell malignancies. Front. Immunol. 2012, 3, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauls, S.D.; Marshall, A.J. Regulation of immune cell signaling by SHIP1: A phosphatase, scaffold protein, and potential therapeutic target. Eur. J. Immunol. 2017, 47, 932–945. [Google Scholar] [CrossRef]
- Ricker, E.; Chowdhury, L.; Yi, W.; Pernis, A.B. The RhoA-ROCK pathway in the regulation of T and B cell responses. F1000Research 2016, 5, 2295. [Google Scholar] [CrossRef] [Green Version]
- Xi, J.; Zhuang, R.; Kong, L.; He, R.; Zhu, H.; Zhang, J. Immunoproteasome-selective inhibitors: An overview of recent developments as potential drugs for hematologic malignancies and autoimmune diseases. Eur. J. Med. Chem. 2019, 182, 111646. [Google Scholar] [CrossRef]
- García-Cuesta, E.M.; Santiago, C.A.; Vallejo-Díaz, J.; Juarranz, Y.; Rodríguez-Frade, J.M.; Mellado, M. The Role of the CXCL12/CXCR4/ACKR3 Axis in Autoimmune Diseases. Front. Endocrinol. (Lausanne) 2019, 10, 585. [Google Scholar] [CrossRef] [Green Version]
- Ulrichts, P.; Guglietta, A.; Dreier, T.; Van Bragt, T.; Hanssens, V.; Hofman, E.; Vankerckhoven, B.; Verheesen, P.; Ongenae, N.; Lykhopiy, V.; et al. Neonatal Fc receptor antagonist efgartigimod safely and sustainably reduces IgGs in humans. J. Clin. Investig. 2018, 128, 4372–4386. [Google Scholar] [CrossRef] [Green Version]
- Tedder, T.F.; Engel, P. CD20: A regulator of cell-cycle progression of B lymphocytes. Immunol. Today 1994, 15, 450–454. [Google Scholar] [CrossRef]
- Horna, P.; Nowakowski, G.; Endell, J.; Boxhammer, R. Comparative Assessment of Surface CD19 and CD20 Expression on B-Cell Lymphomas from Clinical Biopsies: Implications for Targeted Therapies. Blood 2019, 134, 5345. [Google Scholar] [CrossRef]
- Bin Riaz, I.; Zahid, U.; Kamal, M.U.; Husnain, M.; McBride, A.; Hua, A.; Hamadani, A.A.; George, L.; Zeeshan, A.; Sipra, Q.-U.-A.R.; et al. Anti-CD 19 and anti-CD 20 CAR-modified T cells for B-cell malignancies: A systematic review and meta-analysis. Immunother. 2017, 9, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Luo, Y.; Zhu, S.; Wang, X.; Zhao, Y.; Ou, X.; Zhang, T.; Ma, X. The efficacy and safety of anti-CD19/CD20 chimeric antigen receptor- T cells immunotherapy in relapsed or refractory B-cell malignancies:a meta-analysis. BMC Cancer 2018, 18, 929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, G.; Barrett, M.; Foà, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.R.; Spigelman, Z.; Cavacini, L.A.; Posner, M.R. Treatment of Pemphigus Vulgaris with Rituximab and Intravenous Immune Globulin. N. Engl. J. Med. 2006, 355, 1772–1779. [Google Scholar] [CrossRef]
- Joly, P.; Mouquet, H.; Roujeau, J.-C.; D’Incan, M.; Gilbert, D.; Jacquot, S.; Gougeon, M.-L.; Bedane, C.; Muller, R.; Dreno, B.; et al. A Single Cycle of Rituximab for the Treatment of Severe Pemphigus. N. Engl. J. Med. 2007, 357, 545–552. [Google Scholar] [CrossRef]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Geyer, M.B.; Rivière, I.; Sénéchal, B.; Wang, X.; Wang, Y.; Purdon, T.J.; Hsu, M.; Devlin, S.M.; Halton, E.; Lamanna, N.; et al. Autologous CD19-Targeted CAR T Cells in Patients with Residual CLL following Initial Purine Analog-Based Therapy. Mol. Ther. 2018, 26, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Ellebrecht, C.T.; Bhoj, V.G.; Nace, A.; Choi, E.J.; Mao, X.; Cho, M.J.; Di Zenzo, G.; Lanzavecchia, A.; Seykora, J.T.; Cotsarelis, G.; et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science 2016, 353, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.S.; Otani, I.M.; Lax, T.; Hochberg, E.; Banerji, A. Reactions to Rituximab in an Outpatient Infusion Center: A 5-Year Review. J. Allergy Clin. Immunol. Pr. 2017, 5, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Joly, P.; Maho-Vaillant, M.; Prost-Squarcioni, C.; Hebert, V.; Houivet, E.; Calbo, S.; Caillot, F.; Golinski, M.L.; Labeille, B.; Picard-Dahan, C.; et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): A prospective, multicentre, parallel-group, open-label randomised trial. Lancet 2017, 389, 2031–2040. [Google Scholar] [CrossRef]
- Joly, P.; Horwath, B.; Patsatsi, Α.; Uzun, S.; Bech, R.; Beissert, S.; Bergman, R.; Bernard, P.; Borradori, L.; Caproni, M.; et al. Updated S2K guidelines on the management of pemphigus vulgaris and foliaceus initiated by the european academy of dermatology and venereology (EADV). J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Witzig, T.E.; Flinn, I.W.; Gordon, L.I.; Emmanouilides, C.; Czuczman, M.S.; Saleh, M.N.; Cripe, L.; Wiseman, G.; Olejnik, T.; Multani, P.S.; et al. Treatment With Ibritumomab Tiuxetan Radioimmunotherapy in Patients With Rituximab-Refractory Follicular Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2002, 20, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Milani, C.; Mendez-Allwood, D. Ofatumumab, a second-generation anti-CD20 monoclonal antibody, for the treatment of lymphoproliferative and autoimmune disorders. Expert Opin. Investig. Drugs 2009, 18, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Klufas, D.M.; Amerson, E.; Twu, O.; Clark, L.; Shinkai, K. Refractory pemphigus vulgaris successfully treated with ofatumumab. JAAD Case Rep. 2020, 6, 734–736. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.-A.; et al. Blinatumomab for minimal residual disease in adults with B-cell precursor acute lymphoblastic leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [Green Version]
- Jabbour, E.; Ravandi, F.; Kebriaei, P.; Huang, X.; Short, N.J.; Thomas, D.; Sasaki, K.; Rytting, M.; Jain, N.; Konopleva, M.; et al. Salvage Chemoimmunotherapy With Inotuzumab Ozogamicin Combined With Mini-Hyper-CVD for Patients With Relapsed or Refractory Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia: A Phase 2 Clinical Trial. JAMA Oncol. 2018, 4, 230–234. [Google Scholar] [CrossRef]
- Tedder, T.F. CD19: A promising B cell target for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 572–577. [Google Scholar] [CrossRef]
- Stohl, W.; Scholz, J.L.; Cancro, M.P. Targeting BLyS in rheumatic disease: The sometimes-bumpy road from bench to bedside. Curr. Opin. Rheumatol. 2011, 23, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Navarra, S.V.; Guzmán, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.-M.; Thomas, M.; Kim, H.-Y.; León, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Stohl, W.; Furie, R.A.; Lisse, J.R.; McKay, J.D.; Merrill, J.T.; Petri, M.A.; Ginzler, E.M.; Chatham, W.W.; McCune, W.J.; et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum. 2009, 61, 1168–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Weinblatt, M.E.; Van Der Heijde, D.; Rigby, W.F.C.; Van Vollenhoven, R.; Bingham, C.O.; Veenhuizen, M.; Gill, A.; Zhao, F.; Komocsar, W.J.; et al. Efficacy and safety of tabalumab, an anti-B-cell-activating factor monoclonal antibody, in patients with rheumatoid arthritis who had an inadequate response to methotrexate therapy: Results from a phase III multicentre, randomised, double-blind study. Ann. Rheum. Dis. 2015, 74, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Bracewell, C.; Isaacs, J.D.; Emery, P.; Ng, W.-F. Atacicept, a novel B cell-targeting biological therapy for the treatment of rheumatoid arthritis. Expert Opin. Biol. Ther. 2009, 9, 909–919. [Google Scholar] [CrossRef]
- Merrill, J.T.; Wallace, D.J.; Wax, S.; Kao, A.; Fraser, P.A.; Chang, P.; Isenberg, D. Efficacy and Safety of Atacicept in Patients With Systemic Lupus Erythematosus: Results of a Twenty-Four-Week, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Parallel-Arm, Phase IIb Study. Arthritis Rheumatol. (Hoboken NJ) 2018, 70, 266–276. [Google Scholar] [CrossRef] [Green Version]
- Bible, E. Multiple sclerosis: Atacicept increases relapse rates in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 182. [Google Scholar] [CrossRef]
- Matsushita, T.; Kobayashi, T.; Mizumaki, K.; Kano, M.; Sawada, T.; Tennichi, M.; Okamura, A.; Hamaguchi, Y.; Iwakura, Y.; Hasegawa, M.; et al. BAFF inhibition attenuates fibrosis in scleroderma by modulating the regulatory and effector B cell balance. Sci. Adv. 2018, 4, eaas9944. [Google Scholar] [CrossRef] [Green Version]
- Rushworth, S.A.; MacEwan, D.J.; Bowles, K.M. Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 1277–1279. [Google Scholar] [CrossRef]
- Khan, W.N. Regulation of B Lymphocyte Development and Activation by Bruton’s Tyrosine Kinase. Immunol. Res. 2001, 23, 147–156. [Google Scholar] [CrossRef]
- Ng, Y.-S.; Wardemann, H.; Chelnis, J.; Cunningham-Rundles, C.; Meffre, E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 2004, 200, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crofford, L.J.; Nyhoff, L.E.; Sheehan, J.H.; Kendall, P.L. The role of Bruton’s tyrosine kinase in autoimmunity and implications for therapy. Expert Rev. Clin. Immunol. 2016, 12, 763–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutcheson, J.; Vanarsa, K.; Bashmakov, A.; Grewal, S.; Sajitharan, D.; Chang, B.Y.; Buggy, J.J.; Zhou, X.J.; Du, Y.; Satterthwaite, A.B.; et al. Modulating proximal cell signaling by targeting Btk ameliorates humoral autoimmunity and end-organ disease in murine lupus. Arthritis Res. Ther. 2012, 14, R243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okkenhaug, K.; Vanhaesebroeck, B. PI3K in lymphocyte development, differentiation and activation. Nat. Rev. Immunol. 2003, 3, 317–330. [Google Scholar] [CrossRef]
- Okkenhaug, K.; Bilancio, A.; Farjot, G.; Priddle, H.; Sancho, S.; Peskett, E.; Pearce, W.; Meek, S.E.; Salpekar, A.; Waterfield, M.D.; et al. Impaired B and T Cell Antigen Receptor Signaling in p110delta PI 3-Kinase Mutant Mice. Science 2002, 297, 1031–1034. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Carter, R.H.; Soltoff, S.P.; Fearon, D.T. CD19 of B cells as a surrogate kinase insert region to bind phosphatidylinositol 3-kinase. Science 1993, 260, 986–989. [Google Scholar] [CrossRef]
- Reif, K.; Okkenhaug, K.; Sasaki, T.; Penninger, J.M.; Vanhaesebroeck, B.; Cyster, J.G. Cutting Edge: Differential Roles for Phosphoinositide 3-Kinases, p110γ and p110δ, in Lymphocyte Chemotaxis and Homing. J. Immunol. 2004, 173, 2236–2240. [Google Scholar] [CrossRef] [Green Version]
- Henley, T.; Kovesdi, D.; Turner, M. B-cell responses to B-cell activation factor of the TNF family (BAFF) are impaired in the absence of PI3K delta. Eur. J. Immunol. 2008, 38, 3543–3548. [Google Scholar] [CrossRef]
- Martínez, N.; Camacho, F.I.; Algara, P.; Rodríguez, A.; Dopazo, A.; Ruíz-Ballesteros, E.; Martín, P.; Martinez-Climent, J.A.; García-Conde, J.; Menárguez, J.; et al. The molecular signature of mantle cell lymphoma reveals multiple signals favoring cell survival. Cancer Res. 2003, 63, 8226–8232. [Google Scholar]
- Randis, T.M.; Puri, K.D.; Zhou, H.; Diacovo, T.G. Role of PI3Kdelta and PI3Kgamma in inflammatory arthritis and tissue localization of neutrophils. Eur. J. Immunol. 2008, 38, 1215–1224. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; Depinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 Kinase Signals BCR-Dependent Mature B Cell Survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borlado, L.R.; Redondo, C.; Alvarez, B.; Jimenez, C.; Criado, L.M.; Flores, J.; Marcos, M.A.R.; Martinez-A, C.; Balomenos, D.; Carrera, A.C. Increased phosphoinositide 3-kinase activity induces a lymphoproliferative disorder and contributes to tumor generation in vivo. FASEB J. 2000, 14, 895–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, E.; Bardi, G.; Bell, S.E.; Chantry, D.; Downes, C.P.; Gray, A.; Humphries, L.A.; Rawlings, D.; Reynolds, H.; Vigorito, E.; et al. A Crucial Role for the p110δ Subunit of Phosphatidylinositol 3-Kinase in B Cell Development and Activation. J. Exp. Med. 2002, 196, 753–763. [Google Scholar] [CrossRef]
- Barber, D.F.; Bartolomé, A.; Hernandez, C.; Flores, J.M.; Redondo, C.; Fernandez-Arias, C.; Camps, M.; Rückle, T.; Schwarz, M.K.; Rodríguez, S.; et al. PI3Kγ inhibition blocks glomerulonephritis and extends lifespan in a mouse model of systemic lupus. Nat. Med. 2005, 11, 933–935. [Google Scholar] [CrossRef] [PubMed]
- Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B.; et al. Blockade of PI3Kγ suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat. Med. 2005, 11, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Helmer, E.; Watling, M.; Jones, E.; Tytgat, D.; Jones, M.; Allen, R.; Payne, A.; Koch, A.; Healy, E. First-in-human studies of seletalisib, an orally bioavailable small-molecule PI3Kδ inhibitor for the treatment of immune and inflammatory diseases. Eur. J. Clin. Pharmacol. 2017, 73, 581–591. [Google Scholar] [CrossRef] [Green Version]
- Roller, A.; Perino, A.; Dapavo, P.; Soro, E.; Okkenhaug, K.; Hirsch, E.; Ji, H. Blockade of Phosphatidylinositol 3-Kinase (PI3K)δ or PI3Kγ Reduces IL-17 and Ameliorates Imiquimod-Induced Psoriasis-like Dermatitis. J. Immunol. 2012, 189, 4612–4620. [Google Scholar] [CrossRef] [Green Version]
- Chacko, G.W.; Tridandapani, S.; Damen, J.E.; Liu, L.; Krystal, G.; Coggeshall, K.M. Negative signaling in B lymphocytes induces tyrosine phosphorylation of the 145-kDa inositol polyphosphate 5-phosphatase, SHIP. J. Immunol. 1996, 157, 2234–2238. [Google Scholar]
- Saxton, T.M.; Van Oostveen, I.; Bowtell, D.; Aebersold, R.; Gold, M.R. B cell antigen receptor cross-linking induces phosphorylation of the p21ras oncoprotein activators SHC and mSOS1 as well as assembly of complexes containing SHC, GRB-2, mSOS1, and a 145-kDa tyrosine-phosphorylated protein. J. Immunol. 1994, 153, 623–636. [Google Scholar]
- Bolland, S.; Pearse, R.N.; Kurosaki, T.; Ravetch, J.V. SHIP Modulates Immune Receptor Responses by Regulating Membrane Association of Btk. Immunity 1998, 8, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Brauweiler, A.; Tamir, I.; Porto, J.D.; Benschop, R.J.; Helgason, C.D.; Humphries, R.K.; Freed, J.H.; Cambier, J.C. Differential Regulation of B Cell Development, Activation, and Death by the Src Homology 2 Domain–Containing 5′ Inositol Phosphatase (Ship). J. Exp. Med. 2000, 191, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akerlund, J.; Getahun, A.; Cambier, J.C. B cell expression of the SH2-containing inositol 5-phosphatase (SHIP-1) is required to establish anergy to high affinity, proteinacious autoantigens. J. Autoimmun. 2015, 62, 45–54. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.K.; Getahun, A.; Gauld, S.B.; Merrell, K.T.; Tamir, I.; Smith, M.J.; Porto, J.M.D.; Li, Q.-Z.; Cambier, J.C. Monophosphorylation of CD79a and CD79b ITAM Motifs Initiates a SHIP-1 Phosphatase-Mediated Inhibitory Signaling Cascade Required for B Cell Anergy. Immunity 2011, 35, 746–756. [Google Scholar] [CrossRef] [Green Version]
- Taher, T.E.; Parikh, K.; Flores-Borja, F.; Mletzko, S.; Isenberg, D.A.; Peppelenbosch, M.P.; Mageed, R.A. Protein phosphorylation and kinome profiling reveal altered regulation of multiple signaling pathways in B lymphocytes from patients with systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2412–2423. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, X.; Lang, R.A.; Guo, F. RhoA of the Rho Family Small GTPases Is Essential for B Lymphocyte Development. PLoS ONE 2012, 7, e33773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saci, A.; Carpenter, C.L. RhoA GTPase Regulates B Cell Receptor Signaling. Mol. Cell 2005, 17, 205–214. [Google Scholar] [CrossRef]
- Ricker, E.; Chinenov, Y.; Pannellini, T.; Flores-Castro, D.; Ye, C.; Gupta, S.; Manni, M.; Liao, J.K.; Pernis, A.B. Serine-threonine kinase ROCK2 regulates germinal center B cell positioning and cholesterol biosynthesis. J. Clin. Investig. 2020, 130, 3654–3670. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Weiss, J.M.; Trzeciak, A.; Chen, W.; Zhang, J.; Nyuydzefe, M.S.; Arencibia, C.; Polimera, S.; Schueller, O.; Fuentes-Duculan, J.; et al. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J. Immunol. 2017, 198, 3809–3814. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Benoist, C.; Mathis, D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2010, 107, 4658–4663. [Google Scholar] [CrossRef] [Green Version]
- Hiepe, F.; Dörner, T.; Hauser, A.E.; Hoyer, B.F.; Mei, H.; Radbruch, A. Long-lived autoreactive plasma cells drive persistent autoimmune inflammation. Nat. Rev. Rheumatol. 2011, 7, 170–178. [Google Scholar] [CrossRef]
- Bohannon, C.; Powers, R.; Satyabhama, L.; Cui, A.; Tipton, C.; Michaeli, M.; Skountzou, I.; Mittler, R.S.; Kleinstein, S.H.; Mehr, R.; et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat. Commun. 2016, 7, 11826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahévas, M.; Michel, M.; Weill, J.-C.; Reynaud, C.-A. Long-Lived Plasma Cells in Autoimmunity: Lessons from B-Cell Depleting Therapy. Front. Immunol. 2013, 4, 494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, K.; Hayden, P.J.; Will, A.; Wheatley, K.; Coyne, I. Bortezomib for the treatment of multiple myeloma. Cochrane Database Syst. Rev. 2016, 4, CD010816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.; Irwin, D.; Stadtmauer, E.; Facon, T.; Harousseau, J.-L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma: Final time-to-event results of the APEX trial. Blood 2007, 110, 3557–3560. [Google Scholar] [CrossRef] [Green Version]
- Neubert, K.; Meister, S.; Moser, K.; Weisel, F.; Maseda, D.; Amann, K.; Wiethe, C.; Winkler, T.H.; Kalden, J.R.; Manz, R.A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat. Med. 2008, 14, 748–755. [Google Scholar] [CrossRef]
- Gomez, A.M.; Vrolix, K.; Martínez-Martínez, P.; Molenaar, P.C.; Phernambucq, M.; Van Der Esch, E.; Duimel, H.; Verheyen, F.; Voll, R.E.; Manz, R.A.; et al. Proteasome Inhibition with Bortezomib Depletes Plasma Cells and Autoantibodies in Experimental Autoimmune Myasthenia Gravis. J. Immunol. 2011, 186, 2503–2513. [Google Scholar] [CrossRef] [Green Version]
- Alexander, T.; Cheng, Q.; Klotsche, J.; Khodadadi, L.; Waka, A.; Biesen, R.; Hoyer, B.F.; Burmester, G.R.; Radbruch, A.; Hiepe, F. Proteasome inhibition with bortezomib induces a therapeutically relevant depletion of plasma cells in SLE but does not target their precursors. Eur. J. Immunol. 2018, 48, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- Jakez-Ocampo, J.; Atisha-Fregoso, Y.; Llorente, L. Refractory Primary Sjögren Syndrome Successfully Treated With Bortezomib. JCR: J. Clin. Rheumatol. 2015, 21, 31–32. [Google Scholar] [CrossRef]
- Saeed, L.; Schmidt, T.H.; Gensler, L.S.; Gross, A.J.; Fox, L.P.; Scharschmidt, T.C.; Gaensler, K.; Naik, H.; Rosenblum, M.A.; Shinkai, K. Successful treatment of mucous membrane pemphigoid with bortezomib. JAAD Case Rep. 2018, 4, 81–83. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, A. CXCL12/CXCR4 signal transduction in diseases and its molecular approaches in targeted-therapy. Immunol. Lett. 2020, 217, 91–115. [Google Scholar] [CrossRef]
- Cheng, Q.; Khodadadi, L.; Taddeo, A.; Klotsche, J.; Hoyer, B.F.; Radbruch, A.; Hiepe, F. CXCR4–CXCL12 interaction is important for plasma cell homing and survival in NZB/W mice. Eur. J. Immunol. 2018, 48, 1020–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khodadadi, L.; Cheng, Q.; Radbruch, A.; Hiepe, F. The Maintenance of Memory Plasma Cells. Front. Immunol. 2019, 10, 721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiepe, F.; Radbruch, A. Plasma cells as an innovative target in autoimmune disease with renal manifestations. Nat. Rev. Nephrol. 2016, 12, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cuadrón, D.; Boluda, B.; Martínez, P.; Bergua, J.; Rodríguez-Veiga, R.; Esteve, J.; Vives, S.; Serrano, J.; Vidriales, B.; Salamero, O.; et al. A phase I–II study of plerixafor in combination with fludarabine, idarubicin, cytarabine, and G-CSF (PLERIFLAG regimen) for the treatment of patients with the first early-relapsed or refractory acute myeloid leukemia. Ann. Hematol. 2018, 97, 763–772. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.D.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators’ Consortium study (POE 10-03). Pediatr. Blood Cancer 2017, 64, e26414. [Google Scholar] [CrossRef]
- Liu, H.; Liu, H.; Deng, X.; Chen, M.; Han, X.; Yan, W.; Wang, N. CXCR4 antagonist delivery on decellularized skin scaffold facilitates impaired wound healing in diabetic mice by increasing expression of SDF-1 and enhancing migration of CXCR4-positive cells. Wound Repair Regen. 2017, 25, 652–664. [Google Scholar] [CrossRef]
- DiPersio, J.F.; Micallef, I.N.; Stiff, P.J.; Bolwell, B.J.; Maziarz, R.T.; Jacobsen, E.; Nademanee, A.; Mccarty, J.; Bridger, G.; Calandra, G. Phase III Prospective Randomized Double-Blind Placebo-Controlled Trial of Plerixafor Plus Granulocyte Colony-Stimulating Factor Compared With Placebo Plus Granulocyte Colony-Stimulating Factor for Autologous Stem-Cell Mobilization and Transplantation for Patients With Non-Hodgkin’s Lymphoma. J. Clin. Oncol. 2009, 27, 4767–4773. [Google Scholar] [CrossRef]
- D’Alterio, C.; Barbieri, A.; Portella, L.; Palma, G.; Polimeno, M.; Riccio, A.; Ierano, C.; Franco, R.; Scognamiglio, G.; Bryce, J.; et al. Inhibition of stromal CXCR4 impairs development of lung metastases. Cancer Immunol. Immunother. 2012, 61, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Pyzik, M.; Rath, T.; Lencer, W.I.; Baker, K.; Blumberg, R.S. FcRn: The Architect Behind the Immune and Nonimmune Functions of IgG and Albumin. J. Immunol. 2015, 194, 4595–4603. [Google Scholar] [CrossRef] [Green Version]
- Rath, T.; Kuo, T.T.; Baker, K.; Qiao, S.-W.; Kobayashi, K.; Yoshida, M.; Roopenian, D.; Fiebiger, E.; Lencer, W.I.; Blumberg, R.S. The Immunologic Functions of the Neonatal Fc Receptor for IgG. J. Clin. Immunol. 2013, 33 (Suppl. S1), S9–S17. [Google Scholar] [CrossRef]
- Hansen, R.J.; Balthasar, J.P. Intravenous Immunoglobulin Mediates an Increase in Anti-Platelet Antibody Clearance via the FcRn Receptor. Thromb. Haemost. 2002, 88, 898–899. [Google Scholar] [CrossRef] [PubMed]
- Reimann, P.M.; Mason, P.D. Plasmapheresis: Technique and complications. Intensiv. Care Med. 1990, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Yaguchi, H.; Takamori, K.; Ogawa, H. Plasmapheresis for the Treatment of Pemphigus Vulgaris and Bullous Pemphigoid. Ther. Apher. 1997, 1, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Higashihara, T.; Kawase, M.; Kobayashi, M.; Hara, M.; Matsuzaki, H.; Uni, R.; Matsumura, M.; Etoh, T.; Takano, H. Evaluating the Efficacy of Double-Filtration Plasmapheresis in Treating Five Patients With Drug-Resistant Pemphigus. Ther. Apher. Dial. Off. Peer Rev. J. Int. Soc. Apher. Jpn. Soc. Apher. Jpn. Soc. Dial. Ther. 2017, 21, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Yano, C.; Ishiji, T.; Kamide, R.; Niimura, M. A Case of Pemphigus Vulgaris Successfully Treated with Single Filtration Plasmapheresis: A Correlation of Clinical Disease Activity with Serum Antibody Levels. J. Dermatol. 2000, 27, 380–385. [Google Scholar] [CrossRef]
- Eming, R.; Hertl, M. Immunoadsorption in pemphigus. Autoimmun. 2006, 39, 609–616. [Google Scholar] [CrossRef]
- Schmidt, E.; Klinker, E.; Opitz, A.; Herzog, S.; Sitaru, C.; Goebeler, M.; Taleghoni, B.M.; Brocker, E.-B.; Zillikens, D. Protein A immunoadsorption: A novel and effective adjuvant treatment of severe pemphigus. Br. J. Dermatol. 2003, 148, 1222–1229. [Google Scholar] [CrossRef]


{kind=link}
| Target Cell | Target Structure | Mode of Action | Pro | Contra |
|---|---|---|---|---|
| Pro-B-cells–PCs | CD19 [160] e.g., XmAb 5871 (obexelimab) | Antibody-mediated depletion of a broad range of B cells including follicular dendritic cells and PCs to reduce autoantibody formation | Effects on autoreactive PCs and autoantibody production | Depletion of long-lived PCs and thus deprivation of protection provided by vaccination |
| Pre-B-cells–mature B cells | CD20 [161] e.g., MabThera (Rituximab) | Antibody-mediated depletion of peripheral B cells relatively sparing long-lived PCs Reduced production of inflammatory cytokines and activation of T cells | Potent therapeutic effect Biosimilars already available for first-in-class drug Rituximab Subcutaneous administration possible (Ofatumumab) “reset” of B cell compartiment may shift balance towards Bregs | Immunogenicity limits effect and safety in Rituximab Self-reactive B cells in immunologic niches (e.g., BM, TLS) may persist Severe immunosuppression; contraindication for live vaccination |
| Naïve and mature B cells | BLyS/ BAFF [162] e.g., Benlysta (Belimumab) | Antibody or fusion protein mediated reduction in B cell activation and survival of autoimmune B cells via reduced survival factors | Proposed relative selectivity for autoimmune B cells as they depend on overproduction of BLyS Might serve as a steroid sparing agent with good safety and tolerability | Medium potency effects; so far only add on therapy |
| B cells excluding PCs | BTK [163] e.g., Imbruvica (Ibrutinib) | Inhibition of BCR downstream signaling Initiation of apoptosis of aberrant B cells | Orally available Therapy well established in B cell malignancies; much experience More specific second generation inhibitors might be better tolerable | Common side effects limit its use, for Ibrutinib specifically increased risk of bleeding |
| B cells including innate like B cells and B1 cells, (T cells) | PI3Kδ [164] e.g., Zydelig (Idelalisib) | Selective inhibition of the isoform mainly expressed on hematopoietic cells which is crucial for B cell survival and proliferation | Orally available Promising pipeline of numerous compounds | Isoform specificity varies between different drugs Common side effects include skin reactions including severe cutaneous adverse events Paradoxical immune activation has been described as inhibition of T-reg cells is stronger than inhibition of T-eff cells |
| B/T cells, NK cells, mast cells, dendritic cells, macrophages | SHIP1 [165] e.g., AQX-1125 (Rosiptor) | Activation of SHIP1 leads to inhibitory interaction with BCR via different pathways, e.g., downregulation of PI3K signaling | Orally available Well tolerable | Negative clinical trials for other indications derogate expectations Multiplicity of cellular functions incompletely understood |
| B/T cells, other immune cells, non-hematopoietic cells | ROCK2 [166] e.g., KD025 (Belumosudil) | Inhibition leads to down/up regulation of central pro/anti-inflammatory interleukins (IL17/IL10) which leads to a decreased TH17 response and anti-inflammatory regulation of B cell subsets | Orally available Promising pipeline of numerous compounds Postulated beneficial effects on cardiovascular system | Involvement in numerous biological processes makes off-target side effects likely |
| PCs | Proteasome [132] e.g., Velcade (Bortezomib) | Enhanced apoptosis of PCs via disruption of intracellular protein degradation | Orally available Therapy well established in myeloma therapy; much experience Potential to eliminate long-lived PCs | Severe side effects including neuropathy might limit long term use and use in less severe cases |
| Autoreactive PCs | Immuno- Proteasome [167] e.g., KZR-616 | Selective inhibition of inflammatory PC proteasome opposed to wide PC inhibition of first in-class proteasome inhibitors (Bortezomib) | Postulated selectivity | So far, rather experimental approach |
| PCs | CXCR4 [168] e.g., Mozobil (Plerixafor) | Dislocation of aberrant PCs from immune-privileged niches | High level of experience from therapy of hematologic malignancies | Rather theoretical approach for autoimmune disease Mechanistically, monotherapy unlikely to be sufficiently effective |
| (PCs) | IgG/FcRn [169] e.g., ARGX-113 (Efgartigimod) | Neutralization of global IgG including autoantibodies | No impairment of other immunoglobulins and albumin when compared to plasmapheresis Approach appears to be a safe alternative to apparative removal of immunoglobulins | No specificity towards aberrant/self-reactive immunoglobulins |
| (PCs) | Immuno- globulins (plasma- pheresis, immune- adsorption) [147] | Removal of pathogenic autoantibodies via exchange of blood plasma or selective removal of compounds by specific membranes | Well-established add on therapy to other therapeutic approaches High level of experience | Limited availability Invasive and time consuming procedure Severe side effects due to removal of albumins and other plasma proteins (hypoglobulinemia) by plasmapheresis lead to severe immunosuppression |
| Drug Name | Target Structure | Condition | Phase | NCT Identifier |
|---|---|---|---|---|
| XmAb 5871 (Obexelimab) (humanized Fc engineered antibody) | CD19 | SLE | II | NCT02725515 (completed) |
| Rituximab (chimeric antibody) | CD20 First in-class | Bullous pemphigoid | I/II III | NCT00286325 (completed) NCT00525616 (completed) |
| Ocular cicatricial pemphigoid | I/II | NCT00584935 (completed) | ||
| Mucous membrane pemphigoid | III | NCT03295383 (recruiting) | ||
| Pemphigus vulgaris | II/III III II | NCT00213512 (completed) NCT01299857 (completed) NCT04400994 (recruiting; +IVIG) | ||
| Dermatomyositis | II | NCT00106184 (completed) | ||
| Ocrelizumab (humanized antibody) | CD20 | Systemic lupus erythematosus | III | NCT00539838 (terminated) |
| Obinutuzumab (humanized antibody) | CD20 | Chronic GvHD | II | NCT02867384 (recruiting) |
| Veltuzumab (humanized antibody) | CD20 | Pemphigus vulgaris | Case report only | |
| Ofatumumab (fully human antibody) | CD20 | Pemphigus vulgaris | III III | NCT02613910 (terminated) NCT01920477 (terminated) |
| Belimumab (fully human neutralizing antibody) | BLyS (also named BAFF) | CLE | Pooled Analysis III | NCT01858792 (completed) BELI-SKIN: 2017-003051-35 (recruiting) |
| Diffuse cutaneous systemic sclerosis | II | NCT01670565 (completed) | ||
| Atacicept (recombinant fusion protein) | BLyS/APRIL | SLE | II II/III | NCT02070978 (completed) NCT00624338 (completed) |
| A-623 (Blisibimod) (recombinant fusion protein) | BLyS | SLE | II III | NCT01162681 (completed) NCT01395745 (completed) |
| LY2127399 (Tabalumab) (human neutralizing antibody) | BLyS | SLE | III III | NCT02041091 (terminated) NCT01205438 (completed) |
| VAY736 (Ianalumab) (human antibody) | BLyS | Pemphigus vulgaris | II | NCT01930175 (completed) |
| Ibrutinib (oral irreversible inhibitor) | BTK First in-class | Chronic GvHD | III II | NCT02959944 (active, not recruiting) NCT04294641 (recruiting) |
| Acalabrutinib (oral irreversible inhibitor) | BTK | GvHD | II | NCT04198922 (recruiting) |
| PRN1008 (oral reversible inhibitor) | BTK | Pemphigus vulgaris | II III | NCT02704429 (completed) NCT03762265 (recruiting) |
| ABBV-105 (Elsubrutinib) (oral irreversible inhibitor) | BTK | SLE | II II | NCT04451772 (recruiting) NCT03978520 (recruiting) |
| Idelalisib (oral reversible inhibitor) | PI3Kδ First in-class | Allergic rhinitis | I | NCT00836914 (completed) |
| Parsaclisib (oral reversible inhibitor) | PI3Kδ | Pemphigus vulgaris | II | NCT03780166 (withdrawn) |
| UCB-5857 (Seletalisib) (oral reversible inhibitor) | PI3Kδ | Psoriasis vulgaris | I | NCT02303509 (completed) |
| AQX-1125 (Rosiptor) (oral reversible activator) | SHIP1 | Atopic eczema | II | NCT02324972 (completed) |
| KD025 (Belumosudil) (oral inhibitor) | ROCK2 | Diffuse cutaneous systemic sclerosis | II | NCT03919799 (recruiting) |
| Psoriasis vulgaris | II II II | NCT02106195 (completed) NCT02317627 (completed) NCT02852967 (completed) | ||
| ARGX-113/ Efgartigimod (neutralizing human antibody) | IgG/FcRn | Pemphigus vulgaris and foliaceus | II | NCT03334058 (active, not recruiting) |
| SYNT001 (monoclonal IgG4 antibody) | IgG/FcRN | Pemphigus vulgaris and foliaceus | I/II | NCT03075904 (completed) |
| KZR-616 (oral irreversible inhibitor) | Immuno-proteasome | Dermatomyositis | II | NCT04033926 (recruiting) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fetter, T.; Niebel, D.; Braegelmann, C.; Wenzel, J. Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches. Cells 2020, 9, 2627. https://doi.org/10.3390/cells9122627
Fetter T, Niebel D, Braegelmann C, Wenzel J. Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches. Cells. 2020; 9(12):2627. https://doi.org/10.3390/cells9122627
Chicago/Turabian StyleFetter, Tanja, Dennis Niebel, Christine Braegelmann, and Joerg Wenzel. 2020. "Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches" Cells 9, no. 12: 2627. https://doi.org/10.3390/cells9122627
APA StyleFetter, T., Niebel, D., Braegelmann, C., & Wenzel, J. (2020). Skin-Associated B Cells in the Pathogenesis of Cutaneous Autoimmune Diseases—Implications for Therapeutic Approaches. Cells, 9(12), 2627. https://doi.org/10.3390/cells9122627