The Role of gp130 Cytokines in Tuberculosis


Abstract
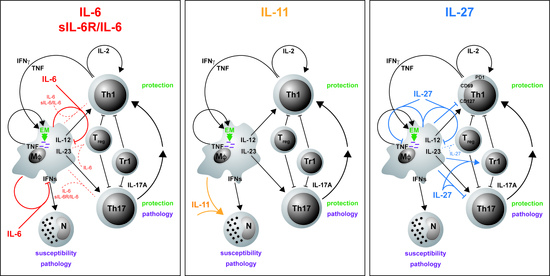
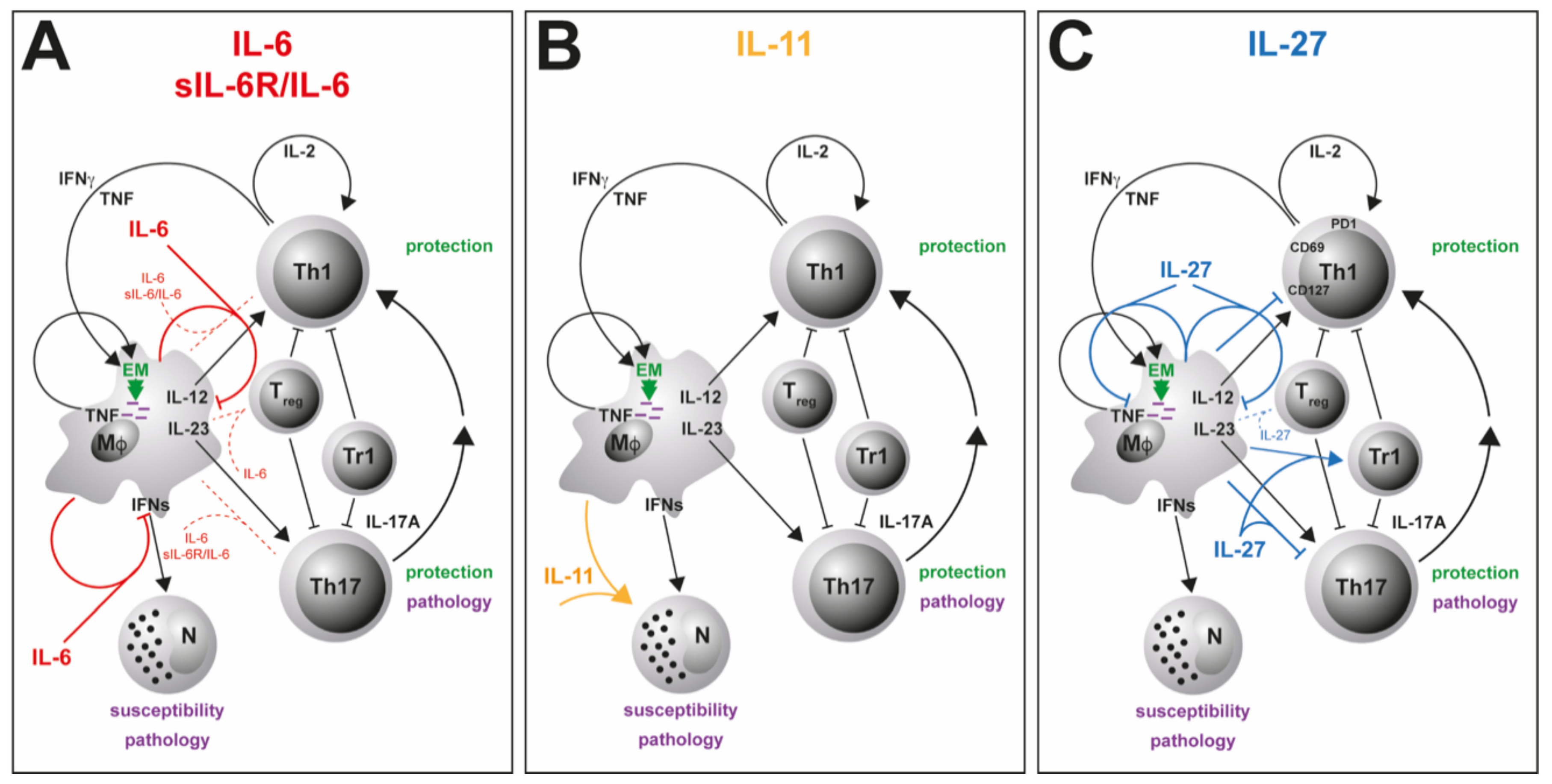
:
1. Introduction
2. gp130-Related Cytokines
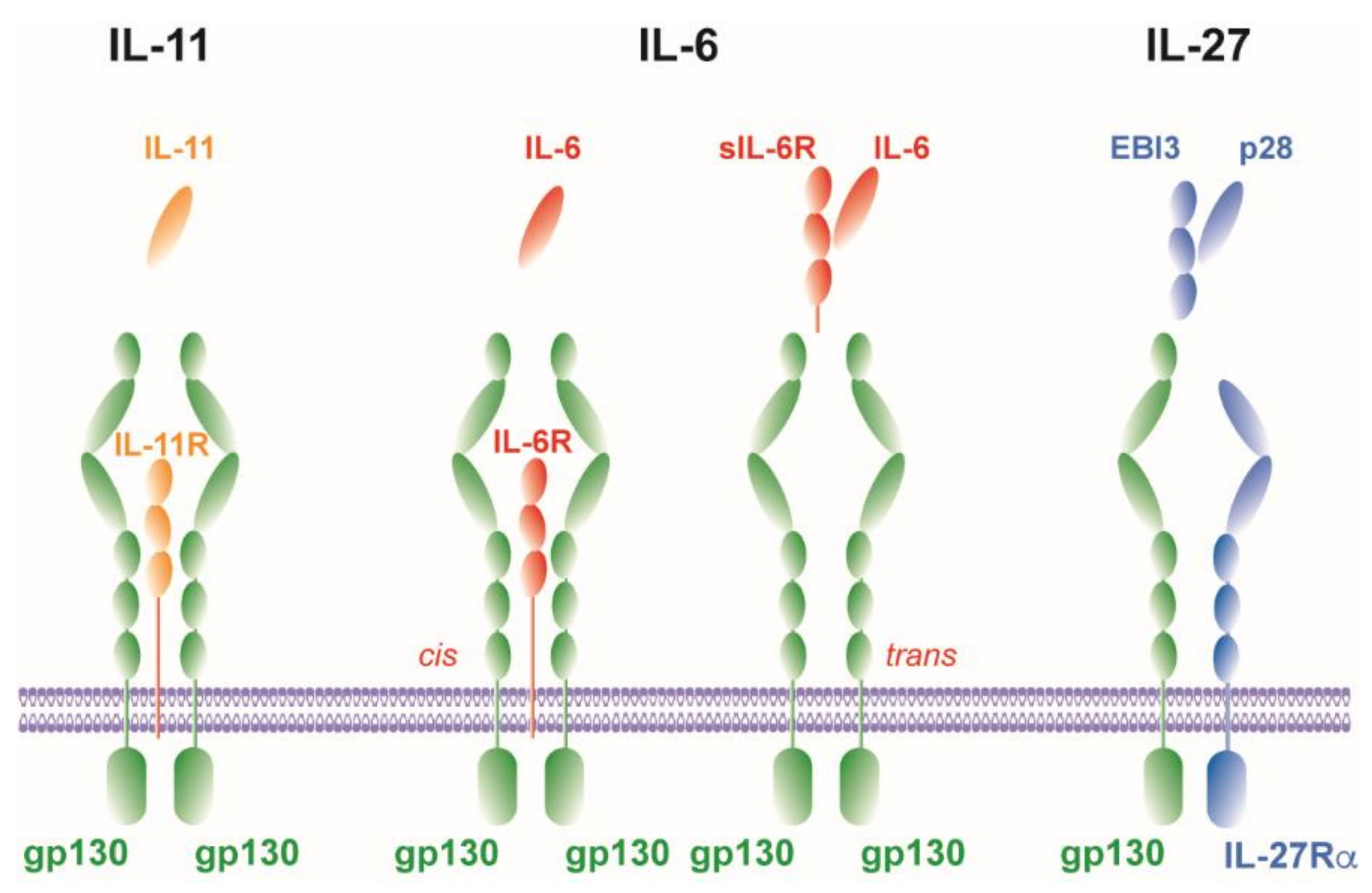
2.1. IL-6
2.1.1. Cis-Signaling
2.1.2. Trans-Signaling
2.1.3. Trans-Presentation
2.2. IL-11
2.3. IL-27
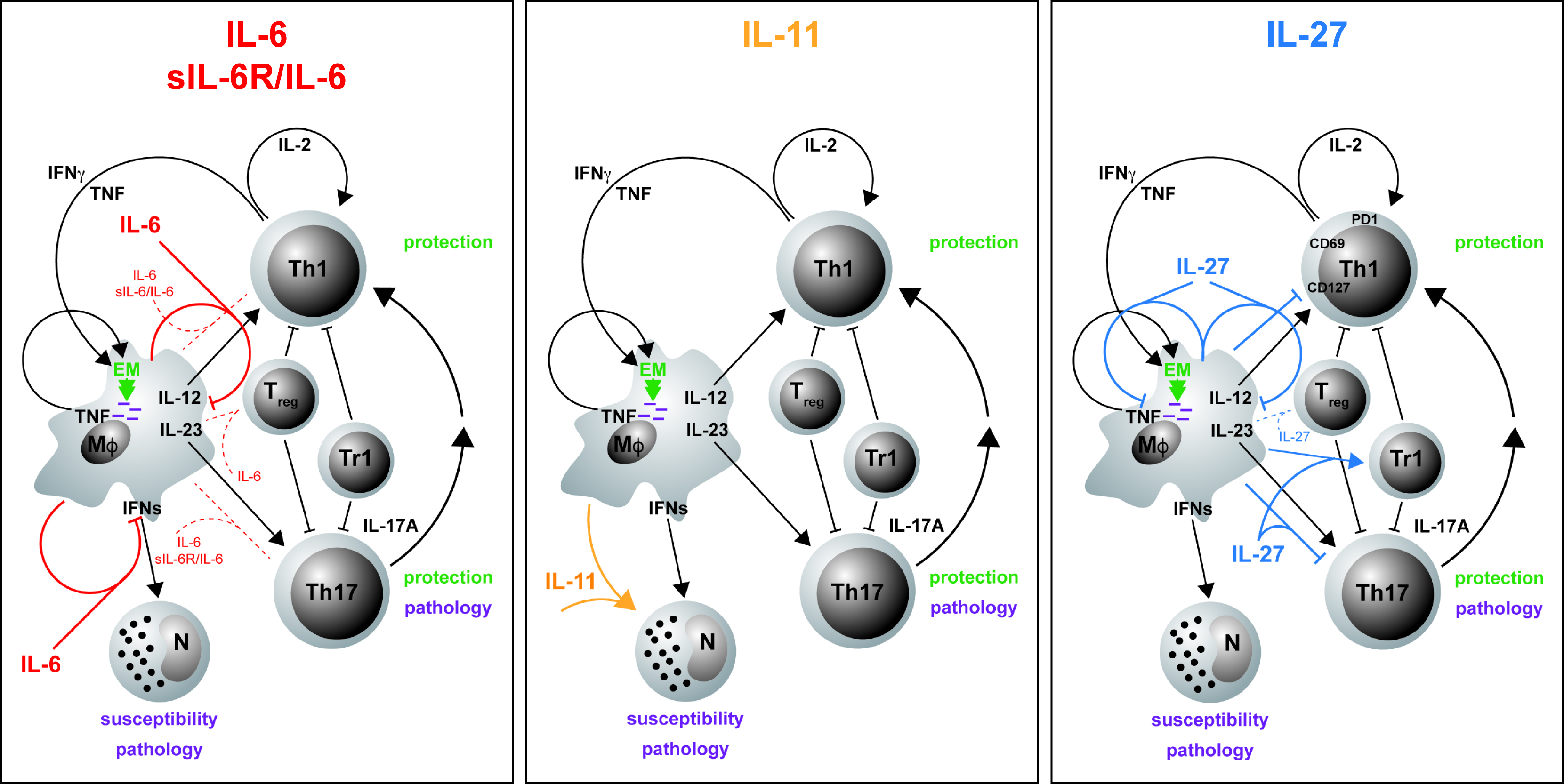
3. gp130 Cytokines in Tuberculosis
3.1. IL-6
3.2. IL-6/sIL-6Rα
3.3. IL-11
3.4. IL-27
3.5. Other gp130 Cytokines
4. Therapeutical Aspects
4.1. Prevention of TB during Therapeutic Targeting of gp130 Cytokines in Autoimmune and Chronic Inflammatory Diseases
4.2. Potential of Targeting gp130 Cytokines during Treatment of TB
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Kaufmann, S.H.E. Protection Against Tuberculosis: Cytokines, T Cells, and Macrophages. Ann. Rheum. Dis. 2002, 6, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, S.; Schaible, U.E. The Granuloma in Tuberculosis: Dynamics of a Host-Pathogen Collusion. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, J. TNF-blocking agents and tuberculosis: New drugs illuminate an old topic. Rheumatology 2005, 44, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, O.N.; Noiseux, C.R.; Martin, J.A.C.; Lara, V.G. Reactivation tuberculosis in a patient with anti-TNF-alpha treatment. Am. J. Gastroenterol. 2001, 96, 1665–1666. [Google Scholar] [CrossRef] [PubMed]
- Sartori, N.S.; de Andrade, N.P.B.; da Silva Chakr, R.M. Incidence of tuberculosis in patients receiving anti-TNF therapy for rheumatic diseases: A systematic review. Clin. Rheumatol. 2020, 39, 1439–1447. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, W.; Yang, G.; Xu, Z.; Wang, J.; Cheng, Q.; Yu, M. Risk of Tuberculosis in Patients Treated with TNF-α Antagonists: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. BMJ Open 2017, 7, e012567. [Google Scholar] [CrossRef] [Green Version]
- Tobin, D.M. Host-Directed Therapies for Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef]
- Hoft, D.; Abate, G. Immunotherapy for tuberculosis: Future prospects. ImmunoTargets Ther. 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Battista Migliori, G. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Dara, Y.; Volcani, D.; Shah, K.; Shin, K.; Venketaraman, V. Potentials of host-directed therapies in tuberculosis management. J. Clin. Med. 2019, 8, 1166. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and chemokines in mycobacterium tuberculosis infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchese, J.M.; Evans, S.; Hult, C.; Joslyn, L.R.; Wessler, T.; Millar, J.A.; Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol. Rev. 2018, 285, 147–167. [Google Scholar] [CrossRef]
- Torrado, E.; Cooper, A.M. Cytokines in the balance of protection and pathology during mycobacterial infections. Adv. Exp. Med. Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Mayer-Barber, K.D.; Sher, A. Role of Innate Cytokines in Mycobacterial Infection. Mucosal Immunol. 2011, 4, 252–260. [Google Scholar] [CrossRef]
- Sharma, S.; Bose, M. Role of Cytokines in Immune Response to Pulmonary Tuberculosis. Asian Pac. J. Allergy Immunol. 2001, 19, 213–219. [Google Scholar]
- Erdmann, H.; Behrends, J.; Ritter, K.; Holscher, A.; Volz, J.; Rosenkrands, I.; Holscher, C. The increased protection and pathology in Mycobacterium tuberculosis-infected IL-27R-alpha-deficient mice is supported by IL-17A and is associated with the IL-17A-induced expansion of multifunctional T cells. Mucosal Immunol. 2018, 11, 1168–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölscher, C.; Hölscher, A.; Rückerl, D.; Yoshimoto, T.; Yoshida, H.; Mak, T.; Saris, C.; Ehlers, S. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J. Immunol. 2005, 174, 3534–3544. [Google Scholar] [CrossRef] [PubMed]
- Van Crevel, R.; Ottenhoff, T.H.M.; Van der Meer, J.W.M. Innate immunity to Mycobacterium tuberculosis. Am. Soc. Microbiol. J. 2002, 15, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.A.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schrelber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Zeng, G.; Zhang, G.; Chen, X. Th1 cytokines, true functional signatures for protective immunity against TB? Chin. Soc. Immunol. 2018, 15, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, E.; Iona, E.; Ferroni, L.; Miettinen, M.; Fattorini, L.; Orefici, G.; Julkunen, I.; Coccia, E.M. Infection of human macrophages and dendritic cells with mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T Cell response. J. Immunol. 2001, 166, 7033–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in Tuberculosis: Friend or Foe. Semin Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon 7 in resistance to mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon gamma gene-distrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [Green Version]
- Newport, M.J.; Huxley, C.M.; Huston, S.; Hawrylowicz, C.M.; Oostra, B.A.; Williamson, R.; Levin, M. A mutation in the interferon-γ–receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 1996, 335, 1941–1949. [Google Scholar] [CrossRef]
- Gallegos, A.M.; van Heijst, J.W.J.; Samstein, M.; Su, X.; Pamer, E.G.; Glickman, M.S. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection In Vivo. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in tuberculosis: Protection, pathology, and biomarkers. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, I.Y.; Panteleev, A.V.; Sosunova, E.V.; Karpina, N.L.; Bagdasarian, T.R.; Burmistrova, I.A.; Andreevskaya, S.N.; Chernousova, L.N.; Vasilyeva, I.A.; Lyadova, I.V. Antigen-Specific IFN-γ Responses correlate with the activity of m. tuberculosis infection but are not associated with the severity of tuberculosis disease. J. Immunol. Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Okamoto Yoshida, Y.; Umemura, M.; Yahagi, A.; O’Brien, R.L.; Ikuta, K.; Kishihara, K.; Hara, H.; Nakae, S.; Iwakura, Y.; Matsuzaki, G. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 2010, 184, 4414–4422. [Google Scholar] [CrossRef] [Green Version]
- Torrado, E.; Cooper, A.M. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010, 21, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, S.A.; Pearl, J.E.; Sakamoto, K.; Gilmartin, L.; Bell, G.K.; Jelley-Gibbs, D.M.; Ghilardi, N.; de Sauvage, F.; Cooper, A.M. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 2005, 175, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, M.; Yahagi, A.; Hamada, S.; Begum, M.D.; Watanabe, H.; Kawakami, K.; Suda, T.; Sudo, K.; Nakae, S.; Iwakura, Y.; et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 2007, 178, 3786–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázár-Molnár, E.; Chen, B.; Sweeney, K.A.; Wang, E.J.; Liu, W.; Lin, J.; Porcelli, S.A.; Almo, S.C.; Nathenson, S.G.; Jacobs, W.R. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13402–13407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott-Browne, J.P.; Shafiani, S.; Tucker-Heard, G.; Ishida-Tsubota, K.; Fontenot, J.D.; Rudensky, A.Y.; Bevan, M.J.; Urdahl, K.B. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J. Exp. Med. 2007, 204, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J., 3rd; Jorg, S.; Heinemann, E.; Oberbeck-Muller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef]
- Lin, P.L.; Plessner, H.L.; Voitenok, N.N.; Flynn, J.A.L. Tumor necrosis factor and tuberculosis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Derouet, D.; Rousseau, F.; Alfonsi, F.; Froger, J.; Hermann, J.; Barbier, F.; Perret, D.; Diveu, C.; Guillet, C.; Preisser, L.; et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc. Natl. Acad Sci. USA 2004, 101, 4827–4832. [Google Scholar] [CrossRef] [Green Version]
- Huyton, T.; Zhang, J.-G.; Luo, C.S.; Lou, M.-Z.; Hilton, D.J.; Nicola, N.A.; Garrett, T.P.J. An unusual cytokine:Ig-domain interaction revealed in the crystal structure of leukemia inhibitory factor (LIF) in complex with the LIF receptor. Proc. Natl. Acad Sci. USA 2007, 104, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelievre, E.; Plun-Favreau, H.; Chevalier, S.; Froger, J.; Guillet, C.; Elson, G.C.; Gauchat, J.F.; Gascan, H. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine: Specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. J. Biol. Chem. 2001, 276, 22476–22484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, D.; He, W.; Sze, K.H.; Gong, K.; Smith, D.K.; Zhu, G.; Ip, N.Y. Solution Structure of the C-terminal Domain of the Ciliary Neurotrophic Factor (CNTF) Receptor and Ligand Free Associations among Components of the CNTF Receptor Complex. J. Biol. Chem. 2003, 278, 23285–23294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalfe, R.D.; Aizel, K.; Zlatic, C.O.; Nguyen, P.M.; Morton, C.J.; Lio, D.S.S.; Cheng, H.-C.; Dobson, R.C.J.; Parker, M.W.; Gooley, P.R.; et al. The structure of the extracellular domains of human interleukin 11 α-receptor reveals mechanisms of cytokine engagement. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [Green Version]
- Mosley, B.; De Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef] [Green Version]
- Pennica, D.; Shaw, K.J.; Swanson, T.A.; Moore, M.W.; Shelton, D.L.; Zioncheck, K.A.; Rosenthal, A.; Taga, T.; Paoni, N.F.; Wood, W.I. Cardiotrophin-1. Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 1995, 270, 10915–10922. [Google Scholar] [CrossRef] [Green Version]
- Pflanz, S.; Hibbert, L.; Mattson, J.; Rosales, R.; Vaisberg, E.; Bazan, J.F.; Phillips, J.H.; McClanahan, T.K.; de Waal Malefyt, R.; Kastelein, R.A. WSX-1 and Glycoprotein 130 Constitute a Signal-Transducing Receptor for IL-27. J. Immunol. 2004, 172, 2225. [Google Scholar] [CrossRef]
- Robledo, O.; Fourcin, M.; Chevalier, S.; Guillet, C.; Auguste, P.; Pouplard-Barthelaix, A.; Pennica, D.; Gascan, H. Signaling of the cardiotrophin-1 receptor. Evidence for a third receptor component. J. Biol. Chem. 1997, 272, 4855–4863. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955. [Google Scholar] [CrossRef]
- Somers, W.; Stahl, M.; Seehra, J.S. 1.9 A crystal structure of interleukin 6: Implications for a novel mode of receptor dimerization and signaling. EMBO J. 1997, 16, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Taga, T.; Kishimoto, T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, Y.; Xiao, H.; Liu, X.; Zhang, Y.; Han, G.; Chen, G.; Hou, C.; Ma, N.; Shen, B.; et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur. J. Immunol. 2016, 46, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 2004, 5, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Putheti, P.; Zhou, Q.; Liu, Q.; Gao, W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008, 19, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Wang, Y.; Zhou, G.; Wang, Y.; Li, X. Review: The Roles and Mechanisms of Glycoprotein 130 Cytokines in the Regulation of Adipocyte Biological Function. Inflammation 2019, 42, 790–798. [Google Scholar] [CrossRef]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef] [Green Version]
- West, N.R. Coordination of Immune-Stroma Crosstalk by IL-6 Family Cytokines. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Silver, J.S.; Hunter, C.A. gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Clemens, J.C.; Schubert, H.L.; Stuckey, J.A.; Fischer, M.W.; Hume, D.M.; Saper, M.A.; Dixon, J.E. Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 1992, 267, 23759–23766. [Google Scholar]
- Hirano, T.; Taga, T.; Nakano, N.; Yasukawa, K.; Kashiwamura, S.; Shimizu, K.; Nakajima, K.; Pyun, K.H.; Kishimoto, T. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc. Natl. Acad. Sci. USA 1985, 82, 5490–5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Snick, J.; Cayphas, S.; Vink, A.; Uyttenhove, C.; Coulie, P.G.; Rubira, M.R.; Simpson, R.J. Purification and NH2-terminal amino acid sequence of a T-cell-derived lymphokine with growth factor activity for B-cell hybridomas. Proc. Natl. Acad. Sci. USA 1986, 83, 9679–9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordan, R.P.; Pumphrey, J.G.; Rudikoff, S. Purification and NH2-terminal sequence of a plasmacytoma growth factor derived from the murine macrophage cell line P388D1. J. Immunol. 1987, 139, 813–817. [Google Scholar] [PubMed]
- Gauldie, J.; Richards, C.; Harnish, D.; Lansdorp, P.; Baumann, H. Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl. Acad. Sci. USA 1987, 84, 7251. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Wong, G.G.; Clark, S.C.; Burakoff, S.J.; Herrmann, S.H. B cell stimulatory factor-2 is involved in the differentiation of cytotoxic T lymphocytes. J. Immunol. 1988, 140, 508. [Google Scholar]
- Kishimoto, T. Factors affecting B-cell growth and differentiation. Annu. Rev. Immunol. 1985, 3, 133–157. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Kohler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Simpson, R.J.; Cheers, C. Role of IL-6 in activation of T cells for acquired cellular resistance to Listeria monocytogenes. J. Immunol. 1994, 152, 5375–5380. [Google Scholar]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Hagenstein, J.; Melderis, S.; Nosko, A.; Warkotsch, M.T.; Richter, J.V.; Ramcke, T.; Herrnstadt, G.R.; Scheller, J.; Yan, I.; Mittrücker, H.-W.; et al. A Novel Role for IL-6 Receptor Classic Signaling: Induction of RORγt(+)Foxp3(+) Tregs with Enhanced Suppressive Capacity. J. Am. Soc. Nephrol. 2019, 30, 1439–1453. [Google Scholar] [CrossRef]
- Harbour, S.N.; DiToro, D.F.; Witte, S.J.; Zindl, C.L.; Gao, M.; Schoeb, T.R.; Jones, G.W.; Jones, S.A.; Hatton, R.D.; Weaver, C.T. TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci. Immunol. 2020, 5, eaaw2262. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity In Vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef]
- Sodenkamp, J.; Behrends, J.; Forster, I.; Muller, W.; Ehlers, S.; Holscher, C. gp130 on macrophages/granulocytes modulates inflammation during experimental tuberculosis. Eur. J. Cell Biol. 2011, 90, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Aderka, D.; Le, J.M.; Vilcek, J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J. Immunol. 1989, 143, 3517–3523. [Google Scholar] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300 (Pt 2), 281–290. [Google Scholar] [CrossRef]
- Mülberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef]
- Mackiewicz, A.; Schooltink, H.; Heinrich, P.C.; Rose-John, S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J. Immunol. 1992, 149, 2021–2027. [Google Scholar]
- Fischer, M.; Goldschmitt, J.; Peschel, C.; Brakenhoff, J.P.; Kallen, K.J.; Wollmer, A.; Grötzinger, J.; Rose-John, S.I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997, 15, 142–145. [Google Scholar] [CrossRef]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and Its Soluble Receptor in Induction of Chemokines and Leukocyte Recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Ebihara, N.; Matsuda, A.; Nakamura, S.; Matsuda, H.; Murakami, A. Role of the IL-6 Classic- and Trans-Signaling Pathways in Corneal Sterile Inflammation and Wound Healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8549–8557. [Google Scholar] [CrossRef]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn disease and experimental colitis In Vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef]
- Dominitzki, S.; Fantini, M.C.; Neufert, C.; Nikolaev, A.; Galle, P.R.; Scheller, J.; Monteleone, G.; Rose-John, S.; Neurath, M.F.; Becker, C. Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J. Immunol. 2007, 179, 2041–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Waetzig, G.H.; Scheller, J.; Grötzinger, J.; Seegert, D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin. Ther. Targets 2007, 11, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Chalaris, A.; May, U.; Waetzig, G.H.; Seegert, D.; Williams, A.S.; Jones, S.A.; Rose-John, S.; Scheller, J. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 2008, 111, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doganci, A.; Sauer, K.; Karwot, R.; Finotto, S. Pathological role of IL-6 in the experimental allergic bronchial asthma in mice. Clin. Rev. Allergy Immunol. 2005, 28, 257–270. [Google Scholar] [CrossRef]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39, 1407–1413. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Putoczki, T.; Ernst, M. More than a sidekick: The IL-6 family cytokine IL-11 links inflammation to cancer. J. Leukoc. Biol. 2010, 88, 1109–1117. [Google Scholar] [CrossRef]
- Du, X.X.; Williams, D.A. Interleukin-11: A multifunctional growth factor derived from the hematopoietic microenvironment. Blood 1994, 83, 2023–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonemura, Y.; Kawakita, M.; Masuda, T.; Fujimoto, K.; Kato, K.; Takatsuki, K. Synergistic effects of interleukin 3 and interleukin 11 on murine megakaryopoiesis in serum-free culture. Exp. Hematol. 1992, 20, 1011–1016. [Google Scholar]
- Jenkins, B.J.; Grail, D.; Inglese, M.; Quilici, C.; Bozinovski, S.; Wong, P.; Ernst, M. Imbalanced gp130-Dependent Signaling in Macrophages Alters Macrophage Colony-Stimulating Factor Responsiveness via Regulation of c-fms Expression. Mol. Cell. Biol. 2004, 24, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redlich, C.A.; Gao, X.; Rockwell, S.; Kelley, M.; Elias, J.A. IL-11 enhances survival and decreases TNF production after radiation-induced thoracic injury. J. Immunol. 1996, 157, 1705–1710. [Google Scholar] [PubMed]
- Trepicchio, W.L.; Bozza, M.; Pedneault, G.; Dorner, A.J. Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J. Immunol. 1996, 157, 3627–3634. [Google Scholar] [PubMed]
- Opal, S.M.; Keith, J.C.; Palardy, J.E.; Parejo, N. Recombinant human interleukin-11 has anti-inflammatory actions yet does not exacerbate systemic Listeria infection. J. Infect. Dis. 2000, 181, 754–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opal, S.M.; Jhung, J.W.; Keith, J.C., Jr.; Palardy, J.E.; Parejo, N.A.; Young, L.D.; Bhattacharjee, A. Recombinant human interleukin-11 in experimental Pseudomonas aeruginosa sepsis in immunocompromised animals. J. Infect. Dis. 1998, 178, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Geba, G.P.; Zheng, T.; Ray, P.; Homer, R.J.; Kuhn, C., 3rd; Flavell, R.A.; Elias, J.A. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J. Clin. Investig. 1996, 98, 2845–2853. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Traber, K.E.; Dimbo, E.L.; Symer, E.M.; Korkmaz, F.T.; Jones, M.R.; Mizgerd, J.P.; Quinton, L.J. Roles of interleukin-11 during acute bacterial pneumonia. PLoS ONE 2019, 14, e0221029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kiapour, N.; Kapoor, S.; Khan, T.; Thamilarasan, M.; Tao, Y.; Cohen, S.; Miller, R.; Sobel, R.A.; Markovic-Plese, S. IL-11 Induces Encephalitogenic Th17 Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2019, 203, 1142–1150. [Google Scholar] [CrossRef]
- Lokau, J.; Nitz, R.; Agthe, M.; Monhasery, N.; Aparicio-Siegmund, S.; Schumacher, N.; Wolf, J.; Möller-Hackbarth, K.; Waetzig, G.H.; Grötzinger, J.; et al. Proteolytic Cleavage Governs Interleukin-11 Trans-signaling. Cell Rep. 2016, 14, 1761–1773. [Google Scholar] [CrossRef] [Green Version]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11, eaar7388. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.L.; Vignali, D.A.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res. 2011, 51, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural Biology of Shared Cytokine Receptors. Annu. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gearing, D.P.; Cosman, D. Homology of the p40 subunit of natural killer cell stimulatory factor (NKSF) with the extracellular domain of the interleukin-6 receptor. Cell 1991, 66, 9–10. [Google Scholar] [CrossRef]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. IL-27, a Heterodimeric Cytokine Composed of EBI3 and p28 Protein, Induces Proliferation of Naive CD4+ T Cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, A.E.; Li, Q.; Xie, L.; Xie, J. Biology of IL-27 and its Role in the Host Immunity against Mycobacterium Tuberculosis. Int. J. Biol. Sci. 2015, 11, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Ghilardi, N.; Li, J.; de Sauvage, F.J. IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 15047–15052. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Hamano, S.; Senaldi, G.; Covey, T.; Faggioni, R.; Mu, S.; Xia, M.; Wakeham, A.C.; Nishina, H.; Potter, J.; et al. WSX-1 Is Required for the Initiation of Th1 Responses and Resistance to L. major Infection. Immunity 2001, 15, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Artis, D.; Johnson, L.M.; Joyce, K.; Saris, C.; Villarino, A.; Hunter, C.A.; Scott, P. Cutting Edge: Early IL-4 Production Governs the Requirement for IL-27-WSX-1 Signaling in the Development of Protective Th1 Cytokine Responses following Leishmania major Infection. J. Immunol. 2004, 172, 4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.; Hibbert, L.; Lieberman, L.; Wilson, E.; Mak, T.; Yoshida, H.; Kastelein, R.A.; Saris, C.; Hunter, C.A. The IL-27R (WSX-1) Is Required to Suppress T Cell Hyperactivity during Infection. Immunity 2003, 19, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Yoshimoto, T.; Yasuda, K.; Mizuguchi, J.; Nakanishi, K. IL-27 Suppresses Th2 Cell Development and Th2 Cytokines Production from Polarized Th2 Cells: A Novel Therapeutic Way for Th2-Mediated Allergic Inflammation. J. Immunol. 2007, 179, 4415. [Google Scholar] [CrossRef] [Green Version]
- Batten, M.; Li, J.; Yi, S.; Kljavin, N.M.; Danilenko, D.M.; Lucas, S.; Lee, J.; de Sauvage, F.J.; Ghilardi, N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17–producing T cells. Nat. Immunol. 2006, 7, 929–936. [Google Scholar] [CrossRef]
- Diveu, C.; McGeachy, M.J.; Boniface, K.; Stumhofer, J.S.; Sathe, M.; Joyce-Shaikh, B.; Chen, Y.; Tato, C.M.; McClanahan, T.K.; de Waal Malefyt, R.; et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J. Immunol. 2009, 182, 5748–5756. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; Kim, D.; Kim, S.; Valentin-Torres, A.; Dvorina, N.; Jang, E.; Nagarajavel, V.; DeSilva, T.M.; Li, X.; Ting, A.H.; et al. Treg-specific IL-27Rα deletion uncovers a key role for IL-27 in Treg function to control autoimmunity. Proc. Natl. Acad. Sci. USA 2017, 114, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Do, J.S.; Visperas, A.; Sanogo, Y.O.; Bechtel, J.J.; Dvorina, N.; Kim, S.; Jang, E.; Stohlman, S.A.; Shen, B.; Fairchild, R.L.; et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal Immunol. 2016, 9, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, A.; Carrier, Y.; Peron, J.P.; Bettelli, E.; Kamanaka, M.; Flavell, R.A.; Kuchroo, V.K.; Oukka, M.; Weiner, H.L. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 2007, 8, 1380–1389. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Pot, C.; Jin, H.; Awasthi, A.; Liu, S.M.; Lai, C.Y.; Madan, R.; Sharpe, A.H.; Karp, C.L.; Miaw, S.C.; Ho, I.C.; et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009, 183, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascanfroni, I.D.; Yeste, A.; Vieira, S.M.; Burns, E.J.; Patel, B.; Sloma, I.; Wu, Y.; Mayo, L.; Ben-Hamo, R.; Efroni, S.; et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 2013, 14, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Ruckerl, D.; Hessmann, M.; Yoshimoto, T.; Ehlers, S.; Holscher, C. Alternatively activated macrophages express the IL-27 receptor alpha chain WSX-1. Immunobiology 2006, 211, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ladel, C.H.; Blum, C.; Dreher, A.; Reifenberg, K.; Kopf, M.; Kaufmann, S.H. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997, 65, 4843–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, B.M.; Frank, A.A.; Orme, I.M.; Cooper, A.M. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect. Immun. 2000, 68, 3322–3326. [Google Scholar] [CrossRef] [Green Version]
- Cheekatla, S.S.; Tripathi, D.; Venkatasubramanian, S.; Nathella, P.K.; Paidipally, P.; Ishibashi, M.; Welch, E.; Tvinnereim, A.R.; Ikebe, M.; Valluri, V.L.; et al. NK-CD11c+ Cell Crosstalk in Diabetes Enhances IL-6-Mediated Inflammation during Mycobacterium tuberculosis Infection. PLoS Pathog. 2016, 12, e1005972. [Google Scholar] [CrossRef]
- Appelberg, R.; Castro, A.G.; Pedrosa, J.; Minoprio, P. Role of interleukin-6 in the induction of protective T cells during mycobacterial infections in mice. Immunology 1994, 82, 361–364. [Google Scholar]
- Ritter, K.; Sodenkamp, J.; Hölscher, A.; Behrends, J.; Holscher, C. IL-6 is not absolutely essential for the development of a TH17 immune response after an aerosol infection with Mycobacterium tuberculosis H37rv. Cells 2020. under review. [Google Scholar]
- Fasnacht, N.; Greweling, M.C.; Bollati-Fogolín, M.; Schippers, A.; Müller, W. T-cell-specific deletion of gp130 renders the highly susceptible IL-10-deficient mouse resistant to intestinal nematode infection. Eur. J. Immunol. 2009, 39, 2173–2183. [Google Scholar] [CrossRef]
- Hatzigeorgiou, D.E.; He, S.; Sobel, J.; Grabstein, K.H.; Hafner, A.; Ho, J.L. IL-6 down-modulates the cytokine-enhanced antileishmanial activity in human macrophages. J. Immunol. 1993, 151, 3682–3692. [Google Scholar]
- Nagabhushanam, V.; Solache, A.; Ting, L.M.; Escaron, C.J.; Zhang, J.Y.; Ernst, J.D. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J. Immunol. 2003, 171, 4750–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanHeyningen, T.K.; Collins, H.L.; Russell, D.G. IL-6 produced by macrophages infected with Mycobacterium species suppresses T cell responses. J. Immunol. 1997, 158, 330–337. [Google Scholar] [PubMed]
- Martinez, A.N.; Mehra, S.; Kaushal, D. Role of interleukin 6 in innate immunity to Mycobacterium tuberculosis infection. J. Infect. Dis. 2013, 207, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Telesca, C.; Angelico, M.; Piccolo, P.; Nosotti, L.; Morrone, A.; Longhi, C.; Carbone, M.; Baiocchi, L. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J. Infect. 2007, 54, e223–e226. [Google Scholar] [CrossRef]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Forster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling In Vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef]
- Lang, R.; Pauleau, A.L.; Parganas, E.; Takahashi, Y.; Mages, J.; Ihle, J.N.; Rutschman, R.; Murray, P.J. SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol. 2003, 4, 546–550. [Google Scholar] [CrossRef]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Carow, B.; Reuschl, A.K.; Gavier-Widen, D.; Jenkins, B.J.; Ernst, M.; Yoshimura, A.; Chambers, B.J.; Rottenberg, M.E. Critical and independent role for SOCS3 in either myeloid or T cells in resistance to Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003442. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, T.; Ehlers, S.; Heitmann, L.; Rausch, A.; Mages, J.; Murray, P.J.; Lang, R.; Holscher, C. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J. Immunol. 2009, 183, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmok, E.; Abad Dar, M.; Behrends, J.; Erdmann, H.; Ruckerl, D.; Endermann, T.; Heitmann, L.; Hessmann, M.; Yoshimura, A.; Rose-John, S.; et al. Suppressor of Cytokine Signaling 3 in Macrophages Prevents Exacerbated Interleukin-6-Dependent Arginase-1 Activity and Early Permissiveness to Experimental Tuberculosis. Front. Immunol. 2017, 8, 1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Stewart, K.N.; Bishop, E.; Marek, C.J.; Kluth, D.C.; Rees, A.J.; Wilson, H.M. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and In Vivo. J. Immunol. 2008, 180, 6270–6278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qualls, J.E.; Neale, G.; Smith, A.M.; Koo, M.S.; DeFreitas, A.A.; Zhang, H.; Kaplan, G.; Watowich, S.S.; Murray, P.J. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci. Signal. 2010, 3, ra62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kasmi, K.C.; Qualls, J.E.; Pesce, J.T.; Smith, A.M.; Thompson, R.W.; Henao-Tamayo, M.; Basaraba, R.J.; Konig, T.; Schleicher, U.; Koo, M.S.; et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 2008, 9, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Heitmann, L.; Abad Dar, M.; Schreiber, T.; Erdmann, H.; Behrends, J.; McKenzie, A.N.; Brombacher, F.; Ehlers, S.; Holscher, C. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J. Pathol. 2014, 234, 338–350. [Google Scholar] [CrossRef] [Green Version]
- el-Ahmady, O.; Mansour, M.; Zoeir, H.; Mansour, O. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Ann. Clin. Biochem. 1997, 34 Pt 2, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Buha, I.; Skodric-Trifunovic, V.; Adzic-Vukicevic, T.; Ilic, A.; Blanka-Protic, A.; Stjepanovic, M.; Andelkovic, M.; Vreca, M.; Milin-Lazovic, J.; Spasovski, V.; et al. Relevance of TNF-alpha, IL-6 and IRAK1 gene expression for assessing disease severity and therapy effects in tuberculosis patients. J. Infect. Dev. Ctries 2019, 13, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Hu, Y.J.; Li, F.G.; Chang, X.J.; Zhang, T.H.; Wang, Z.T. Analysis of Cytokine Levers in Pleural Effusions of Tuberculous Pleurisy and Tuberculous Empyema. Mediat. Inflamm. 2016, 2016, 3068103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, Y.; He, G.; Jiang, X.; Chen, P.; Ouyang, J. Differential diagnosis of tuberculous and malignant pleural effusions: Comparison of the Th1/Th2 cytokine panel, tumor marker panel and chemistry panel. Scand. J. Clin. Lab. Investig. 2020, 80, 265–270. [Google Scholar] [CrossRef]
- Anbarasu, D.; Raja, C.P.; Raja, A. Multiplex analysis of cytokines/chemokines as biomarkers that differentiate healthy contacts from tuberculosis patients in high endemic settings. Cytokine 2013, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Akashi, S.; Nagai, H.; Nagase, H.; Nakamura, H.; Matsui, H.; Hebisawa, A.; Ohta, K. Combined Analysis of IFN-γ, IL-2, IL-5, IL-10, IL-1RA and MCP-1 in QFT Supernatant Is Useful for Distinguishing Active Tuberculosis from Latent Infection. PLoS ONE 2016, 11, e0152483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Manna, M.P.; Orlando, V.; Li Donni, P.; Sireci, G.; Di Carlo, P.; Cascio, A.; Dieli, F.; Caccamo, N. Identification of plasma biomarkers for discrimination between tuberculosis infection/disease and pulmonary non tuberculosis disease. PLoS ONE 2018, 13, e0192664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, A.; Condos, R.; Huie, M.L.; Dawson, R.; Dheda, K.; Bateman, E.; Rom, W.N.; Weiden, M.D. Elevated IP-10 and IL-6 from bronchoalveolar lavage cells are biomarkers of non-cavitary tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 922–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Jiao, L.; Liu, T.; Song, J.; Wang, M.; Liang, L.; Wen, C.; Hu, L.; Qu, W.; Ying, B. No Significant Effects of IL-6 and IL-13 Gene Variants on Tuberculosis Susceptibility in the Chinese Population. DNA Cell Biol. 2020, 39, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. Interleukin-6: Discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S2. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Sodenkamp, J.; Waetzig, G.H.; Scheller, J.; Seegert, D.; Grötzinger, J.; Rose-John, S.; Ehlers, S.; Hölscher, C. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology 2012, 217, 996–1004. [Google Scholar] [CrossRef]
- Kapina, M.A.; Shepelkova, G.S.; Avdeenko, V.G.; Guseva, A.N.; Kondratieva, T.K.; Evstifeev, V.V.; Apt, A.S. Interleukin-11 drives early lung inflammation during Mycobacterium tuberculosis infection in genetically susceptible mice. PLoS ONE 2011, 6, e21878. [Google Scholar] [CrossRef] [Green Version]
- Shepelkova, G.; Evstifeev, V.; Majorov, K.; Bocharova, I.; Apt, A. Therapeutic Effect of Recombinant Mutated Interleukin 11 in the Mouse Model of Tuberculosis. J. Infect. Dis. 2016, 214, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Pearl, J.E.; Khader, S.A.; Solache, A.; Gilmartin, L.; Ghilardi, N.; de Sauvage, F.; Cooper, A.M. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J. Immunol. 2004, 173, 7490–7496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrado, E.; Fountain, J.J.; Liao, M.; Tighe, M.; Reiley, W.W.; Lai, R.P.; Meintjes, G.; Pearl, J.E.; Chen, X.; Zak, D.E.; et al. Interleukin 27R regulates CD4+ T cell phenotype and impacts protective immunity during Mycobacterium tuberculosis infection. J. Exp. Med. 2015, 212, 1449–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Dutta, R.K.; Khan, M.A.; Ishaq, M.; Sharma, K.; Malhotra, H.; Majumdar, S. IL-27 inhibits IFN-γ induced autophagy by concomitant induction of JAK/PI3 K/Akt/mTOR cascade and up-regulation of Mcl-1 in Mycobacterium tuberculosis H37Rv infected macrophages. Int. J. Biochem. Cell Biol. 2014, 55, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jung, J.Y.; Nau, G.J. Interferon-γ, tumor necrosis factor, and interleukin-18 cooperate to control growth of Mycobacterium tuberculosis in human macrophages. Cytokine 2012, 60, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Orlova, M.O.; Majorov, K.B.; Lyadova, I.V.; Eruslanov, E.B.; M’Lan, C.E.; Greenwood, C.M.; Schurr, E.; Apt, A.S. Constitutive differences in gene expression profiles parallel genetic patterns of susceptibility to tuberculosis in mice. Infect. Immun. 2006, 74, 3668–3672. [Google Scholar] [CrossRef] [Green Version]
- Lyadova, I.V.; Tsiganov, E.N.; Kapina, M.A.; Shepelkova, G.S.; Sosunov, V.V.; Radaeva, T.V.; Majorov, K.B.; Shmitova, N.S.; van den Ham, H.J.; Ganusov, V.V.; et al. In mice, tuberculosis progression is associated with intensive inflammatory response and the accumulation of Gr-1 cells in the lungs. PLoS ONE 2010, 5, e10469. [Google Scholar] [CrossRef]
- Underhill-Day, N.; McGovern, L.A.; Karpovich, N.; Mardon, H.J.; Barton, V.A.; Heath, J.K. Functional characterization of W147A: A high-affinity interleukin-11 antagonist. Endocrinology 2003, 144, 3406–3414. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Thiem, S.; Nguyen, P.M.; Eissmann, M.; Putoczki, T.L. Epithelial gp130/Stat3 functions: An intestinal signaling node in health and disease. Semin. Immunol. 2014, 26, 29–37. [Google Scholar] [CrossRef]
- Nahid, P.; Bliven-Sizemore, E.; Jarlsberg, L.G.; De Groote, M.A.; Johnson, J.L.; Muzanyi, G.; Engle, M.; Weiner, M.; Janjic, N.; Sterling, D.G.; et al. Aptamer-based proteomic signature of intensive phase treatment response in pulmonary tuberculosis. Tuberculosis (Edinb.) 2014, 94, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Larousserie, F.; Pflanz, S.; Coulomb-L’Herminé, A.; Brousse, N.; Kastelein, R.; Devergne, O. Expression of IL-27 in human Th1-associated granulomatous diseases. J. Pathol. 2004, 202, 164–171. [Google Scholar] [CrossRef]
- Wu, Y.B.; Ye, Z.J.; Qin, S.M.; Wu, C.; Chen, Y.Q.; Shi, H.Z. Combined detections of interleukin 27, interferon-γ, and adenosine deaminase in pleural effusion for diagnosis of tuberculous pleurisy. Chin. Med. J. (Engl.) 2013, 126, 3215–3221. [Google Scholar] [PubMed]
- Yang, W.B.; Liang, Q.L.; Ye, Z.J.; Niu, C.M.; Ma, W.L.; Xiong, X.Z.; Du, R.H.; Zhou, Q.; Zhang, J.C.; Shi, H.Z. Cell origins and diagnostic accuracy of interleukin 27 in pleural effusions. PLoS ONE 2012, 7, e40450. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ye, Z.J.; Zhou, Q.; You, W.J.; Cui, A.; Wang, X.J.; Zhai, K.; Jin, X.G.; Tong, Z.H.; Shi, H.Z. IL-27 and IL-27-producing CD4+ T cells in human tuberculous pleural effusion. Tuberculosis (Edinb.) 2014, 94, 579–588. [Google Scholar] [CrossRef]
- Urazova, O.I.; Churina, E.G.; Hasanova, R.R.; Novitskiy, V.V.; Poletika, V.S. Association between polymorphisms of cytokine genes and secretion of IL-12p70, IL-18, and IL-27 by dendritic cells in patients with pulmonary tuberculosis. Tuberculosis (Edinb.) 2019, 115, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Saunders, B.M.; Britton, W.J. Life and death in the granuloma: Immunopathology of tuberculosis. Immunol. Cell Biol. 2007, 85, 103–111. [Google Scholar] [CrossRef]
- Reiley, W.W.; Shafiani, S.; Wittmer, S.T.; Tucker-Heard, G.; Moon, J.J.; Jenkins, M.K.; Urdahl, K.B.; Winslow, G.M.; Woodland, D.L. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 2010, 107, 19408–19413. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, C.; Hoang, T.T.; Izzo, A.; Billeskov, R.; Troudt, J.; Arnett, K.; Keyser, A.; Elvang, T.; Andersen, P.; Dietrich, J. Protection and polyfunctional T cells induced by Ag85B-TB10.4/IC31 against Mycobacterium tuberculosis is highly dependent on the antigen dose. PLoS ONE 2009, 4, e5930. [Google Scholar] [CrossRef]
- Derrick, S.C.; Yabe, I.M.; Yang, A.; Morris, S.L. Vaccine-induced anti-tuberculosis protective immunity in mice correlates with the magnitude and quality of multifunctional CD4 T cells. Vaccine 2011, 29, 2902–2909. [Google Scholar] [CrossRef]
- Lindenstrøm, T.; Agger, E.M.; Korsholm, K.S.; Darrah, P.A.; Aagaard, C.; Seder, R.A.; Rosenkrands, I.; Andersen, P. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J. Immunol. 2009, 182, 8047–8055. [Google Scholar] [CrossRef]
- Beamer, G.L.; Flaherty, D.K.; Assogba, B.D.; Stromberg, P.; Gonzalez-Juarrero, M.; de Waal Malefyt, R.; Vesosky, B.; Turner, J. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J. Immunol. 2008, 181, 5545–5550. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.; Gonzalez-Juarrero, M.; Ellis, D.L.; Basaraba, R.J.; Kipnis, A.; Orme, I.M.; Cooper, A.M. In Vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 2002, 169, 6343–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.C.; Ciric, B.; Touil, T.; Harle, H.; Grammatikopolou, J.; Das Sarma, J.; Gran, B.; Zhang, G.X.; Rostami, A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 3268–3275. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Ghoreschi, K.; Yang, X.P.; Takahashi, H.; Laurence, A.; Vahedi, G.; Sciumè, G.; Hall, A.O.; Dupont, C.D.; Francisco, L.M.; et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity 2012, 36, 1017–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; O’Dee, D.; Hamilton, T.; Nau, G.J. Cytokines involved in interferon-gamma production by human macrophages. J. Innate Immun. 2010, 2, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Ghaffari, A.A.; Cheng, G. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 2010, 185, 6599–6607. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.Y.; Robinson, C.M. IL-12 and IL-27 regulate the phagolysosomal pathway in mycobacteria-infected human macrophages. Cell Commun. Signal. 2014, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kane, C.M.; Elkington, P.T.; Friedland, J.S. Monocyte-dependent oncostatin M and TNF-alpha synergize to stimulate unopposed matrix metalloproteinase-1/3 secretion from human lung fibroblasts in tuberculosis. Eur. J. Immunol. 2008, 38, 1321–1330. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Kübler, A.; Luna, B.; Larsson, C.; Ammerman, N.C.; Andrade, B.B.; Orandle, M.; Bock, K.W.; Xu, Z.; Bagci, U.; Mollura, D.J.; et al. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J. Pathol. 2015, 235, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Ou, Q.; Wu, J.; Zhang, B.; Shen, L.; Chen, S.; Weng, X.; Zhang, Y.; Zhang, W.; Shao, L. Potential diagnostic value of serum/pleural fluid IL-31 levels for tuberculous pleural effusion. Sci. Rep. 2016, 6, 20607. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Fan, Y.; Shen, D.; Yu, M.; Shi, L.; Ding, S.; Li, L. Characterization of cytokine profile to distinguish latent tuberculosis from active tuberculosis and healthy controls. Cytokine 2020, 135, 155218. [Google Scholar] [CrossRef] [PubMed]
- German Clinical Trials Register. A multi-centre, exploratory trial to assess the mechanisms of molecular activity, safety and tolerability of one dose level of FE 999301 by intravenous infusions in patients with active inflammatory bowel disease (IBD). German Clinical Trials Register 2016. Available online: http://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00010101 (accessed on 15 December 2020).
- I-Mab Biopharma HongKong Limited. Safety and efficacy of TJ301 IV in participants with active ulcerative colitis. U.S. Natl. Libary Med. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03235752 (accessed on 15 December 2020).
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Bijl, H.; Woody, J.N. Repeated therapy with monoclonal antibody to tumour necrosis factor alpha (cA2) in patients with rheumatoid arthritis. Lancet 1994, 344, 1125–1127. [Google Scholar] [CrossRef]
- Mohan, V.P.; Scanga, C.A.; Yu, K.; Scott, H.M.; Tanaka, K.E.; Tsang, E.; Tsai, M.M.; Flynn, J.L.; Chan, J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: Possible role for limiting pathology. Infect. Immun. 2001, 69, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Scanga, C.A.; Mohan, V.P.; Joseph, H.; Yu, K.; Chan, J.; Flynn, J.L. Reactivation of latent tuberculosis: Variations on the Cornell murine model. Infect. Immun. 1999, 67, 4531–4538. [Google Scholar] [CrossRef] [Green Version]
- Afif, W.; Loftus, E.V., Jr. Safety profile of IBD therapeutics: Infectious risks. Gastroenterol. Clin. N. Am. 2009, 38, 691–709. [Google Scholar] [CrossRef]
- Solovic, I.; Sester, M.; Gomez-Reino, J.J.; Rieder, H.L.; Ehlers, S.; Milburn, H.J.; Kampmann, B.; Hellmich, B.; Groves, R.; Schreiber, S.; et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: A TBNET consensus statement. Eur. Respir. J. 2010, 36, 1185–1206. [Google Scholar] [CrossRef] [Green Version]
- Stallmach, A.; Hagel, S.; Bruns, T. Adverse effects of biologics used for treating IBD. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 167–182. [Google Scholar] [CrossRef]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert. Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef]
- Akioka, S. Interleukin-6 in juvenile idiopathic arthritis. Mod. Rheumatol. 2019, 29, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kita, Y.; Kanamaru, N.; Hashimoto, S.; Uchiyama, Y.; Mihara, M.; Inoue, Y.; Ohsugi, Y.; Kishimoto, T.; Sakatani, M. Anti-IL-6 receptor antibody causes less promotion of tuberculosis infection than anti-TNF-alpha antibody in mice. Clin. Dev. Immunol. 2011, 2011, 404929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strand, V.; Ahadieh, S.; French, J.; Geier, J.; Krishnaswami, S.; Menon, S.; Checchio, T.; Tensfeldt, T.G.; Hoffman, E.; Riese, R.; et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res. Ther. 2015, 17, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Arthritis Res. Ther. 2011, 13, R141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Ito, K.; Yamanaka, H. Postmarketing surveillance of tocilizumab for rheumatoid arthritis in Japan: Interim analysis of 3881 patients. Ann. Rheum. Dis. 2011, 70, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Sano, Y.; Yaguramaki, H.; et al. Effectiveness and safety of tocilizumab: Postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J. Rheumatol. 2014, 41, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Prystaz, K.; Kaiser, K.; Kovtun, A.; Haffner-Luntzer, M.; Fischer, V.; Rapp, A.E.; Liedert, A.; Strauss, G.; Waetzig, G.H.; Rose-John, S.; et al. Distinct Effects of IL-6 Classic and Trans-Signaling in Bone Fracture Healing. Am. J. Pathol. 2018, 188, 474–490. [Google Scholar] [CrossRef] [Green Version]
- Hoge, J.; Yan, I.; Jänner, N.; Schumacher, V.; Chalaris, A.; Steinmetz, O.M.; Engel, D.R.; Scheller, J.; Rose-John, S.; Mittrücker, H.W. IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J. Immunol. 2013, 190, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug. Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Ndlovu, H.; Marakalala, M.J. Granulomas and Inflammation: Host-Directed Therapies for Tuberculosis. Front. Immunol. 2016, 7, 434. [Google Scholar] [CrossRef] [Green Version]
- Palucci, I.; Delogu, G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy 2018, 63, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Parida, S.K.; Keshavjee, S.; Cassell, G.; Wallis, R.; Axelsson-Robertsson, R.; Doherty, M.; Andersson, J.; Maeurer, M. Inflammation and tuberculosis: Host-directed therapies. J. Intern. Med. 2015, 277, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aige, C.; Bishai, W.R. Penitentiary or penthouse condo: The tuberculous granuloma from the microbe’s point of view. Cell Microbiol. 2010, 12, 301–309. [Google Scholar] [CrossRef]
- Dietrich, C.; Candon, S.; Ruemmele, F.M.; Devergne, O. A soluble form of IL-27Ralpha is a natural IL-27 antagonist. J. Immunol. 2014, 192, 5382–5389. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, S.; Tubbe, I.; Galle, P.R.; Schild, H.J.; Birkenbach, M.; Blumberg, R.S.; Neurath, M.F. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J. Exp. Med. 2006, 203, 1875–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Receptor Complex | Reference 1 |
|---|---|---|
| IL-6 | IL-6Rα:gp130:gp130 | [50,51] |
| IL-11 | IL11Rα:gp130:gp130 | [45] |
| IL-27 (p28/EBI3) | IL27Rα (WSX-1):gp130 | [48] |
| IL-35 (p35/EBI3) | IL-12Rβ2:gp130 | [40] |
| gp130:gp130 | ||
| IL-39 (p19/EBI3) | IL-23R:gp130 | [53] |
| LIF | LIFR:gp130 | [42] |
| OSM | OSMR:gp130 | [46] |
| LIFR:gp130 | ||
| CNTF | LIFR:CNTFRα:gp130 | [44] |
| LIFR:IL-6Rα:gp130 | ||
| LIFR:sIL-6Rα:gp130 | ||
| CT-1 | LIFR:gp130 | [47,49] |
| LIFR:gp190:gp130 | ||
| CLC | LIFR:mCNTFRα:gp130 | [43] |
| NP | LIFR:CNTFRα:gp130 | [41] |
| Effect 2 | Protection 3 | Pathology 4 | |
|---|---|---|---|
| IL-6 | → inflammation [139] | → [139] | → [139] |
| ↓ macrophages [82,142,143] | |||
| IL-6/sIL-6Rα | → inflammation [169] | → [93] | → [93] |
| IL-11 | ↑ inflammation [170,171] | ↓ [170,171] | ↑ [170,171] |
| ↑ macrophages [171] | |||
| ↑ neutrophils [170,171] | |||
| IL-27 | ↓ inflammation [17,18,172] | ↓ [17,18,172] | ↓ [17,18,172] |
| ↓ CD4 [17,173] | |||
| ↑ Tr1 [17] | |||
| ↓ TH17 [17] | |||
| ↓ macrophages [18,174,175] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritter, K.; Rousseau, J.; Hölscher, C. The Role of gp130 Cytokines in Tuberculosis. Cells 2020, 9, 2695. https://doi.org/10.3390/cells9122695
Ritter K, Rousseau J, Hölscher C. The Role of gp130 Cytokines in Tuberculosis. Cells. 2020; 9(12):2695. https://doi.org/10.3390/cells9122695
Chicago/Turabian StyleRitter, Kristina, Jasmin Rousseau, and Christoph Hölscher. 2020. "The Role of gp130 Cytokines in Tuberculosis" Cells 9, no. 12: 2695. https://doi.org/10.3390/cells9122695
APA StyleRitter, K., Rousseau, J., & Hölscher, C. (2020). The Role of gp130 Cytokines in Tuberculosis. Cells, 9(12), 2695. https://doi.org/10.3390/cells9122695