Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death




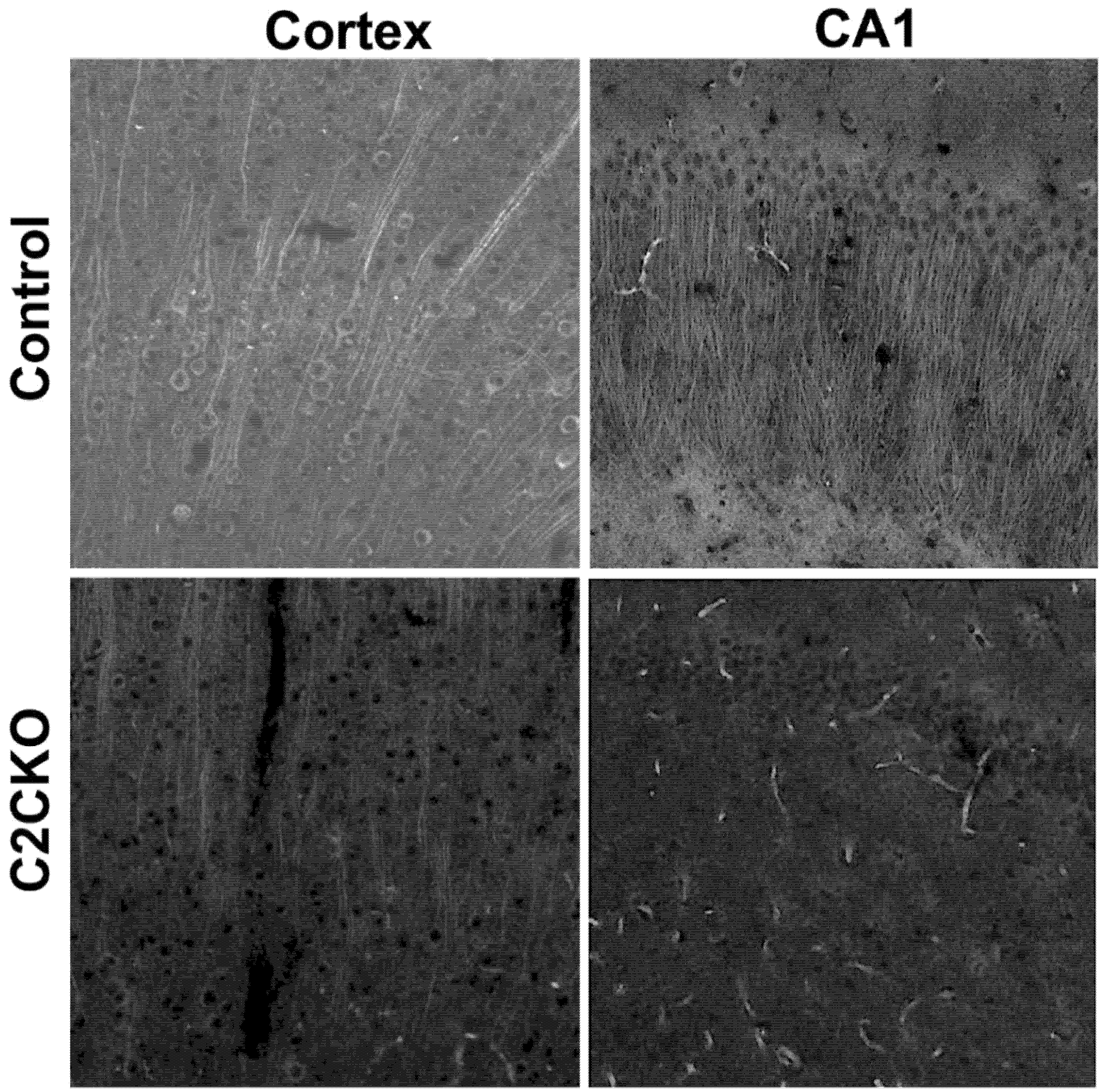{kind=link}
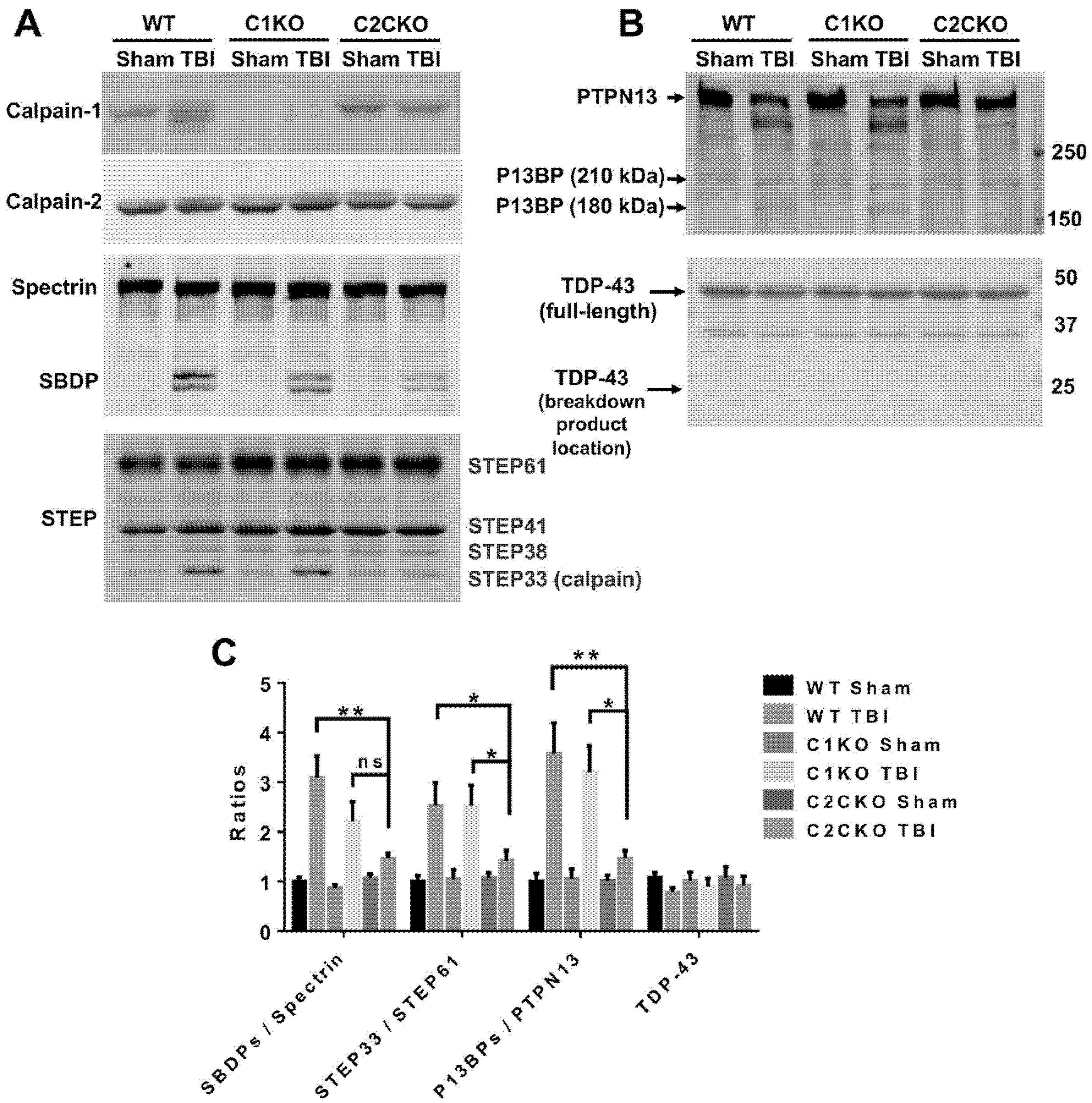{kind=link}
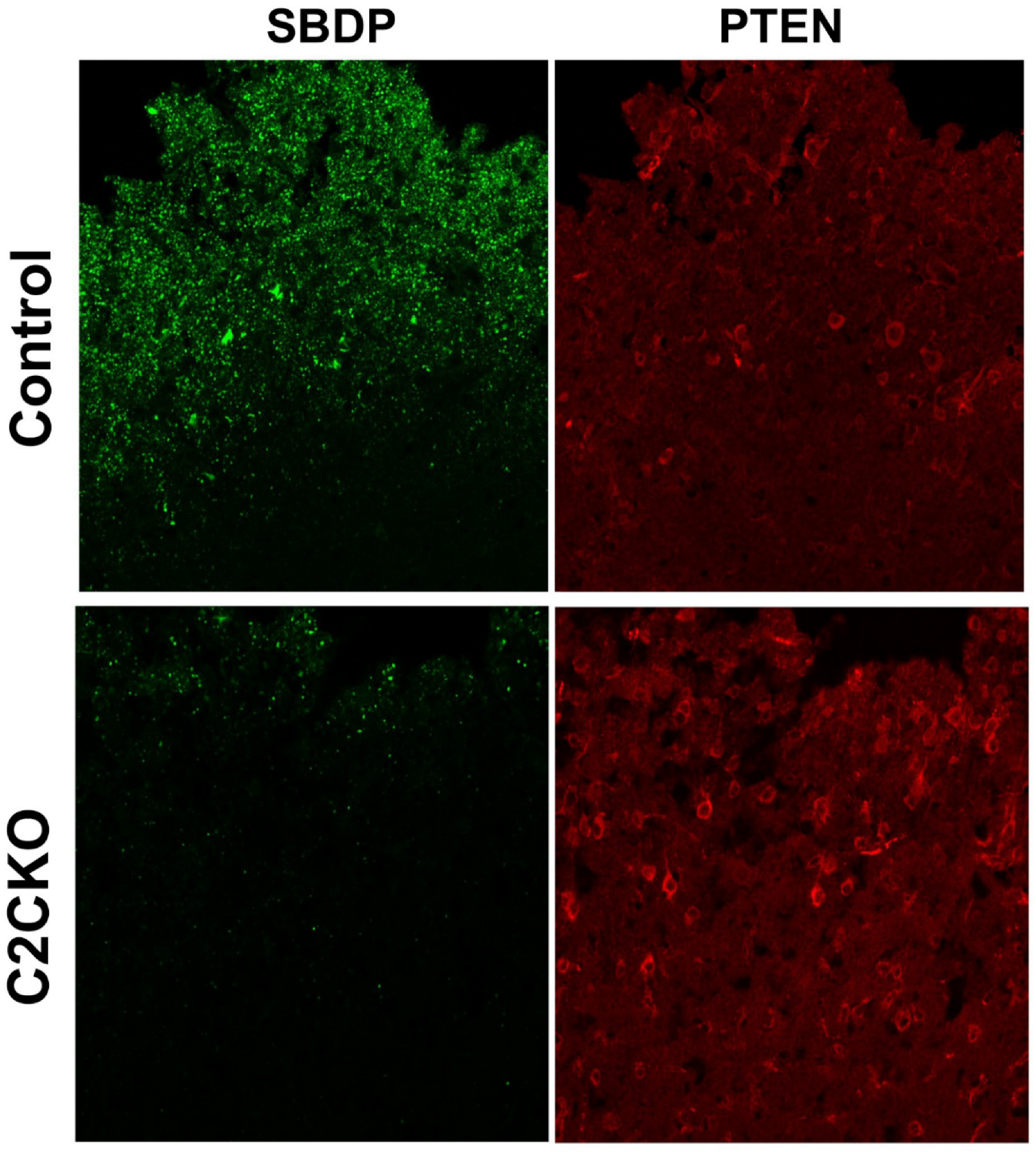{kind=link}
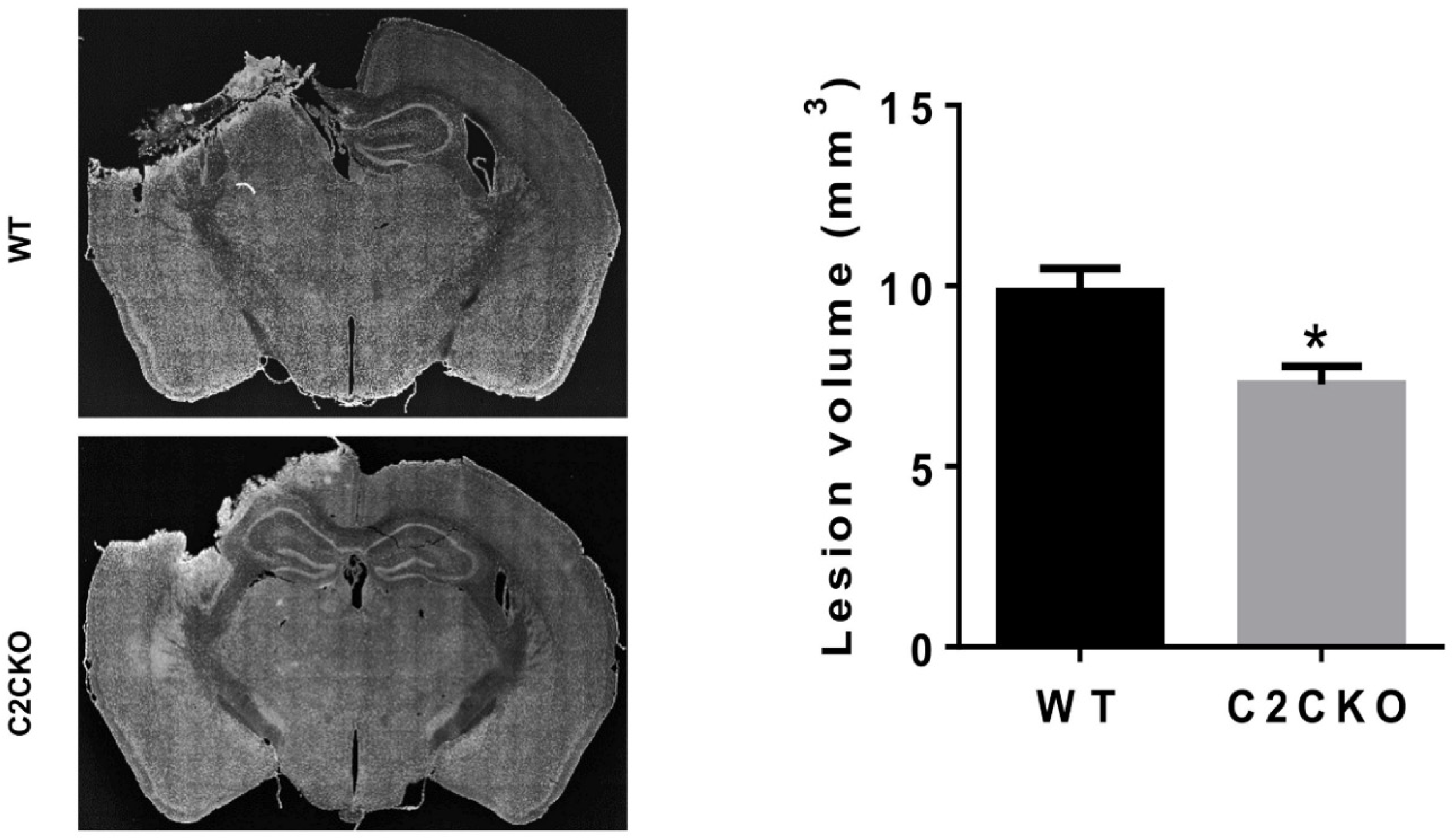{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Calpain-1 Role in Synaptic Plasticity
3. Calpain-1 Role in Neuroprotection
4. Calpain-2 Role in Synaptic Plasticity
5. Calpain-2 Role in Neuronal Death
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guroff, G. A neutral, calcium-activated proteinase from the soluble fraction of rat brain. J. Biol. Chem. 1964, 239, 149–155. [Google Scholar] [PubMed]
- Murachi, T.; Tanaka, K.; Hatanaka, M.; Murakami, T. Intracellular Ca2+-dependent protease (calpain) and its high-molecular-weight endogenous inhibitor (calpastatin). Adv. Enzym. Regul. 1981, 19, 407–424. [Google Scholar] [CrossRef]
- Lynch, G.; Baudry, M. The biochemistry of memory—A new and specific hypothesis. Science 1984, 224, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Amini, M.; Ma, C.-L.; Farazifard, R.; Zhu, G.; Zhang, Y.; Vanderluit, J.; Zoltewicz, J.S.; Hage, F.; Savitt, J.M.; Lagace, D.C. Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP, and promotes neuronal survival following injury. J. Neurosci. 2013, 33, 5773–5784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhu, G.; Briz, V.; Hsu, Y.T.; Bi, X.; Baudry, M. A molecular brake controls the magnitude of long-term potentiation. Nat. Commun. 2014, 5, 3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.; Liu, Y.; Wang, Y.; Bi, X.; Baudry, M. Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J. Neurosci. 2015, 35, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Saito, K.I.; Grynspan, F.; Griffin, W.R.; Katayama, S.; Honda, T.; Mohan, P.S.; Shea, T.B.; Beermann, M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1994, 747, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Vanderklish, P.W.; Bahr, B.A. The pathogenic activation of calpain: A marker and mediator of cellular toxicity and disease states. Int. J. Exp. Pathol. 2000, 81, 323–339. [Google Scholar] [CrossRef]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Camins, A.; Verdaguer, E.; Folch, J.; Pallàs, M. Involvement of calpain activation in neurodegenerative processes. CNS Drug Rev. 2006, 12, 135–148. [Google Scholar] [CrossRef]
- Bevers, M.B.; Neumar, R.W. Mechanistic role of calpains in postischemic neurodegeneration. J. Cereb. Blood Flow Metab. 2008, 28, 655–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vosler, P.; Brennan, C.; Chen, J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, M.C.; Wang, K. Calpain in the CNS: From synaptic function to neurotoxicity. Sci. Signal. 2008, 1, re1. [Google Scholar] [CrossRef]
- Baudry, M.; Bi, X. Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 2016, 39, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Sorimachi, H.; Ono, Y. Regulation and physiological roles of the calpain system in muscular disorders. Cardiovasc. Res. 2012, 96, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macqueen, D.J.; Wilcox, A.H. Characterization of the definitive classical calpain family of vertebrates using phylogenetic, evolutionary and expression analyses. Open Biol. 2014, 4, 130219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Wang, K.K. The calpain family and human disease. Trends Mol. Med. 2001, 7, 355–362. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Bushby, K.M. Advances in the molecular genetics of the limb-girdle type of autosomal recessive progressive muscular dystrophy. Curr. Opin. Neurol. 1996, 9, 389. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, E.; de Luna, N.; Diaz-Manera, J.; Rojas-García, R.; Gonzalez-Quereda, L.; Flix, B.; de Morrée, A.; van der Maarel, S.; Illa, I. Comparison of dysferlin expression in human skeletal muscle with that in monocytes for the diagnosis of dysferlin myopathy. PLoS ONE 2011, 6, e29061. [Google Scholar] [CrossRef]
- Harris, F.; Biswas, S.; Singh, J.; Dennison, S.; Phoenix, D.A. Calpains and their multiple roles in diabetes mellitus. Ann. N. Y. Acad. Sci. 2006, 1084, 452–480. [Google Scholar] [CrossRef]
- Litosh, V.A.; Rochman, M.; Rymer, J.K.; Porollo, A.; Kottyan, L.C.; Rothenberg, M.E. Calpain-14 and its association with eosinophilic esophagitis. J. Allergy Clin. Immunol. 2017, 139, 1762–1771. [Google Scholar] [CrossRef]
- Mahajan, V.B.; Skeie, J.M.; Bassuk, A.G.; Fingert, J.H.; Braun, T.A.; Daggett, H.T.; Folk, J.C.; Sheffield, V.C.; Stone, E.M. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS Genet. 2012, 8, e1003001. [Google Scholar] [CrossRef]
- Zha, C.; Abi Farah, C.; Fonov, V.; Holt, R.; Ceroni, F.; Ragges, N.; Rudko, D.; Sossin, W.S. Disruption of Capn15 in mice leads to brain and eye deficits. bioRxiv 2019, 763888. [Google Scholar] [CrossRef] [Green Version]
- Lugli, G.; Larson, J.; Martone, M.E.; Jones, Y.; Smalheiser, N.R. Dicer and eIF2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J. Neurochem. 2005, 94, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Phan, T.; Mansuy, I.M.; Storm, D.R. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell 2007, 128, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, M.G. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev. Neurosci. 2006, 17, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Pieri, M.; Ciotti, M.T.; Carunchio, I.; Canu, N.; Calissano, P.; Zona, C.; Severini, C. Substance P provides neuroprotection in cerebellar granule cells through Akt and MAPK/Erk activation: Evidence for the involvement of the delayed rectifier potassium current. Neuropharmacology 2007, 52, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- del Cerro, S.; Arai, A.; Kessler, M.; Bahr, B.A.; Vanderklish, P.; Rivera, S.; Lynch, G. Stimulation of NMDA receptors activates calpain in cultured hippocampal slices. Neurosci. Lett. 1994, 167, 149–152. [Google Scholar] [CrossRef]
- Abe, K.; Takeichi, M. NMDA-receptor activation induces calpain-mediated β-catenin cleavages for triggering gene expression. Neuron 2007, 53, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Rong, Y.; Baudry, M. Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci. Lett. 2000, 286, 149–153. [Google Scholar] [CrossRef]
- Lu, X.; Wyszynski, M.; Sheng, M.; Baudry, M. Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: Implications for synaptic plasticity. J. Neurochem. 2001, 77, 1553–1560. [Google Scholar] [CrossRef] [Green Version]
- Jourdi, H.; Lu, X.; Yanagihara, T.; Lauterborn, J.C.; Bi, X.; Gall, C.M.; Baudry, M. Prolonged positive modulation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors induces calpain-mediated PSD-95/Dlg/ZO-1 protein degradation and AMPA receptor down-regulation in cultured hippocampal slices. J. Pharmacol. Exp. Ther. 2005, 314, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-Y.; Lynch, D.R. Calpain and synaptic function. Mol. Neurobiol. 2006, 33, 215–236. [Google Scholar] [CrossRef]
- Khoutorsky, A.; Yanagiya, A.; Gkogkas, C.G.; Fabian, M.R.; Prager-Khoutorsky, M.; Cao, R.; Gamache, K.; Bouthiette, F.; Parsyan, A.; Sorge, R.E. Control of synaptic plasticity and memory via suppression of poly (A)-binding protein. Neuron 2013, 78, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudry, M.; Bi, X.; Gall, C.; Lynch, G. The biochemistry of memory: The 26 year journey of a ‘new and specific hypothesis’. Neurobiol. Learn. Mem. 2011, 95, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burridge, K.; Wennerberg, K. Rho and Rac take center stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.Y.; Rex, C.S.; Casale, M.S.; Gall, C.M.; Lynch, G. Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 2007, 27, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Chen, L.Y.; Sharma, A.; Liu, J.; Babayan, A.H.; Gall, C.M.; Lynch, G. Different Rho GTPase–dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol. 2009, 186, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Briz, V.; Zhu, G.; Wang, Y.; Liu, Y.; Avetisyan, M.; Bi, X.; Baudry, M. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J. Neurosci. 2015, 35, 2269–2282. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Briz, V.; Seinfeld, J.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 deletion impairs mGluR-dependent LTD and fear memory extinction. Sci. Rep. 2017, 7, 42788. [Google Scholar] [CrossRef]
- Heysieattalab, S.; Lee, K.-H.; Liu, Y.; Wang, Y.; Foy, M.R.; Bi, X.; Baudry, M. Impaired cerebellar plasticity and eye-blink conditioning in calpain-1 knock-out mice. Neurobiol. Learn. Mem. 2019, 170, 106995. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Kakegawa, W.; Matsuda, S.; Kohda, K.; Nishiyama, J.; Takahashi, T.; Yuzaki, M. Cerebellar long-term depression requires dephosphorylation of TARP in Purkinje cells. Eur. J. Neurosci. 2012, 35, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Rostamiani, K.; Hsu, Y.-T.; Wang, Y.; Bi, X.; Baudry, M. Calpain-mediated regulation of stargazin in adult rat brain. Neuroscience 2011, 178, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, J.H. Cerebellar learning mechanisms. Brain Res. 2015, 1621, 260–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Van Beugen, B.J.; De Zeeuw, C.I. Distributed synergistic plasticity and cerebellar learning. Nat. Rev. Neurosci. 2012, 13, 619–635. [Google Scholar] [CrossRef] [PubMed]
- Grasselli, G.; Hansel, C. Cerebellar long-term potentiation: Cellular mechanisms and role in learning. In International Review of Neurobiology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 117, pp. 39–51. [Google Scholar]
- Schonewille, M.; Gao, Z.; Boele, H.-J.; Veloz, M.F.V.; Amerika, W.E.; Šimek, A.A.; De Jeu, M.T.; Steinberg, J.P.; Takamiya, K.; Hoebeek, F.E. Reevaluating the role of LTD in cerebellar motor learning. Neuron 2011, 70, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Briz, V.; Chishti, A.; Bi, X.; Baudry, M. Distinct roles for mu-calpain and m-calpain in synaptic NMDAR-mediated neuroprotection and extrasynaptic NMDAR-mediated neurodegeneration. J. Neurosci. 2013, 33, 18880–18892. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hersheson, J.; Lopez, D.; Hammer, M.; Liu, Y.; Lee, K.H.; Pinto, V.; Seinfeld, J.; Wiethoff, S.; Sun, J.; et al. Defects in the CAPN1 Gene Result in Alterations in Cerebellar Development and Cerebellar Ataxia in Mice and Humans. Cell Rep. 2016, 16, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Shetty, A.; Gan-Or, Z.; Ashtiani, S.; Ruskey, J.A.; van de Warrenburg, B.; Wassenberg, T.; Kamsteeg, E.-J.; Rouleau, G.A.; Suchowersky, O. CAPN1 mutations: Expanding the CAPN1-related phenotype: From hereditary spastic paraparesis to spastic ataxia. Eur. J. Med Genet. 2019, 62, 103605. [Google Scholar] [CrossRef]
- Forman, O.P.; De Risio, L.; Mellersh, C.S. Missense mutation in CAPN1 is associated with spinocerebellar ataxia in the Parson Russell Terrier dog breed. PLoS ONE 2013, 8, e64627. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Lopez, D.; Lee, M.; Dayal, S.; Hurtado, A.; Bi, X.; Baudry, M. Protection against TBI-induced neuronal death with post-treatment with a selective calpain-2 inhibitor in mice. J. Neurotrauma 2017, 35, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lopez, D.; Davey, P.G.; Cameron, D.J.; Nguyen, K.; Tran, J.; Marquez, E.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and calpain-2 play opposite roles in retinal ganglion cell degeneration induced by retinal ischemia/reperfusion injury. Neurobiol. Dis. 2016, 93, 121–128. [Google Scholar] [CrossRef]
- Seinfeld, J.; Baudry, N.; Xu, X.; Bi, X.; Baudry, M. Differential activation of calpain-1 and calpain-2 following kainate-induced seizure activity in rats and mice. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Gan-Or, Z.; Bouslam, N.; Birouk, N.; Lissouba, A.; Chambers, D.B.; Vérièpe, J.; Androschuk, A.; Laurent, S.B.; Rochefort, D.; Spiegelman, D. Mutations in CAPN1 cause autosomal-recessive hereditary spastic paraplegia. Am. J. Hum. Genet. 2016, 98, 1038–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, G.; Afonso, P.M.; Salazar, I.L.; Duarte, C.B. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015, 1621, 82–101. [Google Scholar] [CrossRef] [PubMed]
- Zadran, S.; Jourdi, H.; Rostamiani, K.; Qin, Q.; Bi, X.; Baudry, M. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J. Neurosci. 2010, 30, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Leal, G.; Comprido, D.; Duarte, C.B. BDNF-induced local protein synthesis and synaptic plasticity. Neuropharmacology 2014, 76, 639–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briz, V.; Hsu, Y.-T.; Li, Y.; Lee, E.; Bi, X.; Baudry, M. Calpain-2-mediated PTEN degradation contributes to BDNF-induced stimulation of dendritic protein synthesis. J. Neurosci. 2013, 33, 4317–4328. [Google Scholar] [CrossRef] [Green Version]
- Oliver, M.W.; Baudry, M.; Lynch, G. The protease inhibitor leupeptin interferes with the development of LTP in hippocampal slices. Brain Res. 1989, 505, 233–238. [Google Scholar] [CrossRef]
- Denny, J.B.; Polan-Curtain, J.; Rodriguez, S.; Wayner, M.J.; Armstrong, D.L. Evidence that protein kinase M does not maintain long-term potentiation. Brain Res. 1990, 534, 201–208. [Google Scholar] [CrossRef]
- Panja, D.; Bramham, C.R. BDNF mechanisms in late LTP formation: A synthesis and breakdown. Neuropharmacology 2014, 76, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Lin, C.-Y.; Kramár, E.A.; Chen, L.Y.; Gall, C.M.; Lynch, G. Brain-derived neurotrophic factor promotes long-term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 2007, 27, 3017–3029. [Google Scholar] [CrossRef] [PubMed]
- Bevers, M.B.; Lawrence, E.; Maronski, M.; Starr, N.; Amesquita, M.; Neumar, R.W. Knockdown of m-calpain increases survival of primary hippocampal neurons following NMDA excitotoxicity. J. Neurochem. 2009, 108, 1237–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papouin, T.; Oliet, S.H. Organization, control and function of extrasynaptic NMDA receptors. Philos. Trans. R. Soc. B 2014, 369, 20130601. [Google Scholar] [CrossRef] [PubMed]
- Krapivinsky, G.; Krapivinsky, L.; Manasian, Y.; Ivanov, A.; Tyzio, R.; Pellegrino, C.; Ben-Ari, Y.; Clapham, D.E.; Medina, I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 2003, 40, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Kurup, P.; Zhang, Y.; Goebel-Goody, S.M.; Wu, P.H.; Hawasli, A.H.; Baum, M.L.; Bibb, J.A.; Lombroso, P.J. Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J. Neurosci. 2009, 29, 9330–9343. [Google Scholar] [CrossRef]
- Gladding, C.M.; Sepers, M.D.; Xu, J.; Zhang, L.Y.; Milnerwood, A.J.; Lombroso, P.J.; Raymond, L.A. Calpain and STriatal-Enriched protein tyrosine phosphatase (STEP) activation contribute to extrasynaptic NMDA receptor localization in a Huntington’s disease mouse model. Hum. Mol. Genet. 2012, 21, 3739–3752. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, Z.; Deng, W.; Fu, S.; Zhang, C.; Chen, M.; Ju, W.; Wang, D.; He, X. Calpain 2-mediated autophagy defect increases susceptibility of fatty livers to ischemia–reperfusion injury. Cell Death Dis. 2016, 7, e2186. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Sarkar, S.; Cuddon, P.; Ttofi, E.K.; Saiki, S.; Siddiqi, F.H.; Jahreiss, L.; Fleming, A.; Pask, D.; Goldsmith, P. Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol. 2008, 4, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, F.; Das, D.; Kommaddi, R.P.; Diwakar, L.; Gowaikar, R.; Rupanagudi, K.V.; Bennett, D.A.; Ravindranath, V. Isoform-specific hyperactivation of calpain-2 occurs presymptomatically at the synapse in Alzheimer’s disease mice and correlates with memory deficits in human subjects. Sci. Rep. 2018, 8, 13119. [Google Scholar] [CrossRef] [PubMed]
- Gordy, C.; He, Y.-W. The crosstalk between autophagy and apoptosis: Where does this lead? Protein Cell 2012, 3, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; Panda, P.K.; Sinha, N.; Das, D.N.; Bhutia, S.K. Autophagy and apoptosis: Where do they meet? Apoptosis 2014, 19, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Fimia, G.M.; Piacentini, M. Dismantling the autophagic arsenal when it is time to die: Concerted AMBRA1 degradation by caspases and calpains. Autophagy 2012, 8, 1255–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwood, S.M.; Yaqoob, M.M.; Allen, D.A. Caspase and calpain function in cell death: Bridging the gap between apoptosis and necrosis. Ann. Clin. Biochem. 2005, 42, 415–431. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hall, R.A.; Lee, M.; Kamgar-parsi, A.; Bi, X.; Baudry, M. The tyrosine phosphatase PTPN13/FAP-1 links calpain-2, TBI and tau tyrosine phosphorylation. Sci. Rep. 2017, 7, 11771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.S.; Lee, E.H.; Chung, C.W.; Jung, Y.K.; Jin, B.K.; Kim, S.U.; Oh, T.H.; Saido, T.C.; Oh, Y.J. Cleavage of Bax is mediated by caspase-dependent or-independent calpain activation in dopaminergic neuronal cells: Protective role of Bcl-2. J. Neurochem. 2001, 77, 1531–1541. [Google Scholar] [CrossRef] [PubMed]
- Takano, J.; Tomioka, M.; Tsubuki, S.; Higuchi, M.; Iwata, N.; Itohara, S.; Maki, M.; Saido, T.C. Calpain Mediates Excitotoxic DNA Fragmentation via Mitochondrial Pathways in Adult Brains evidence from calpastatin mutant mice. J. Biol. Chem. 2005, 280, 16175–16184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.K. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef]
- Ruiz-Vela, A.; de Buitrago, G.G.; Martínez-A, C. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999, 18, 4988–4998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Wong, T.P.; Chery, N.; Gaertner, T.; Wang, Y.T.; Baudry, M. Calpain-mediated mGluR1α truncation: A key step in excitotoxicity. Neuron 2007, 53, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampfl, A.; Posmantur, R.; Zhao, X.; Schmutzhard, E.; Clifton, G.; Hayes, R. Mechanisms of calpain proteolysis following traumatic brain injury: Implications for pathology and therapy: A review and update. J. Neurotrauma 1997, 14, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.K.; Larner, S.F.; Robinson, G.; Hayes, R.L. Neuroprotection targets after traumatic brain injury. Curr. Opin. Neurol. 2006, 19, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Yin, F.; Zhang, J.; Qian, Y. The role of calpains in traumatic brain injury. Brain Inj. 2014, 28, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Pike, B.R.; Zhao, X.; Newcomb, J.K.; Posmantur, R.M.; Wang, K.K.; Hayes, R.L. Regional calpain and caspase-3 proteolysis of α-spectrin after traumatic brain injury. Neuroreport 1998, 9, 2437–2442. [Google Scholar] [CrossRef]
- Pike, B.R.; Flint, J.; Dutta, S.; Johnson, E.; Wang, K.K.; Hayes, R.L. Accumulation of non-erythroid αII-spectrin and calpain-cleaved αII-spectrin breakdown products in cerebrospinal fluid after traumatic brain injury in rats. J. Neurochem. 2001, 78, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Pineda, J.A.; Lewis, S.B.; Valadka, A.B.; Papa, L.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Aikman, J.M.; Akle, V. Clinical significance of α II-spectrin breakdown products in cerebrospinal fluid after severe traumatic brain injury. J. Neurotrauma 2007, 24, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Brophy, G.M.; Pineda, J.A.; Papa, L.; Lewis, S.B.; Valadka, A.B.; Hannay, H.J.; Heaton, S.C.; Demery, J.A.; Liu, M.C.; Tepas, J.J., III. αII-Spectrin breakdown product cerebrospinal fluid exposure metrics suggest differences in cellular injury mechanisms after severe traumatic brain injury. J. Neurotrauma 2009, 26, 471–479. [Google Scholar] [CrossRef] [Green Version]
- Mondello, S.; Robicsek, S.A.; Gabrielli, A.; Brophy, G.M.; Papa, L.; Tepas, J., III; Robertson, C.; Buki, A.; Scharf, D.; Jixiang, M. αII-spectrin breakdown products (SBDPs): Diagnosis and outcome in severe traumatic brain injury patients. J. Neurotrauma 2010, 27, 1203–1213. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Giovannone, N.; Hanten, G.; Wilde, E.A.; McCauley, S.R.; Hunter, J.V.; Li, X.; Levin, H.S.; Smith, D.H. Evidence That the Blood Biomarker SNTF Predicts Brain Imaging Changes and Persistent Cognitive Dysfunction in Mild TBI Patients. Front. Neurol. 2013, 4, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siman, R.; Shahim, P.; Tegner, Y.; Blennow, K.; Zetterberg, H.; Smith, D.H. Serum SNTF increases in concussed professional ice hockey players and relates to the severity of postconcussion symptoms. J. Neurotrauma 2015, 32, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Siman, R.; Cui, H.; Wewerka, S.; Hamel, L.; Smith, D.H.; Zwank, M. Serum SNTF, a Surrogate Marker of Axonal Injury, Is Prognostic for Lasting Brain Dysfunction in Mild TBI Treated in the Emergency Department. Front. Neurol. 2020, 11, 249. [Google Scholar] [CrossRef] [PubMed]
- Saatman, K.E.; Murai, H.; Bartus, R.T.; Smith, D.H.; Hayward, N.J.; Perri, B.R.; McIntosh, T.K. Calpain inhibitor AK295 attenuates motor and cognitive deficits following experimental brain injury in the rat. Proc. Natl. Acad. Sci. USA 1996, 93, 3428–3433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posmantur, R.; Kampfl, A.; Siman, R.; Liu, S.; Zhao, X.; Clifton, G.; Hayes, R. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience 1997, 77, 875–888. [Google Scholar] [CrossRef]
- Thompson, S.N.; Carrico, K.M.; Mustafa, A.G.; Bains, M.; Hall, E.D. A pharmacological analysis of the neuroprotective efficacy of the brain- and cell-permeable calpain inhibitor MDL-28170 in the mouse controlled cortical impact traumatic brain injury model. J. Neurotrauma 2010, 27, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Bains, M.; Cebak, J.E.; Gilmer, L.K.; Barnes, C.C.; Thompson, S.N.; Geddes, J.W.; Hall, E.D. Pharmacological analysis of the cortical neuronal cytoskeletal protective efficacy of the calpain inhibitor SNJ-1945 in a mouse traumatic brain injury model. J. Neurochem. 2013, 125, 125–132. [Google Scholar] [CrossRef]
- Seubert, P.; Baudry, M.; Dudek, S.; Lynch, G. Calmodulin stimulates the degradation of brain spectrin by calpain. Synapse 1987, 1, 20–24. [Google Scholar] [CrossRef]
- Yamashita, T.; Hideyama, T.; Hachiga, K.; Teramoto, S.; Takano, J.; Iwata, N.; Saido, T.C.; Kwak, S. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat. Commun. 2012, 3, 1307. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Nham, A.; Sherbaf, A.; Quach, D.; Yahya, E.; Ranburger, D.; Bi, X.; Baudry, M. Calpain-2 as a therapeutic target in repeated concussion–induced neuropathy and behavioral impairment. Sci. Adv. 2020, 6, eaba5547. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. https://doi.org/10.3390/cells9122698
Wang Y, Liu Y, Bi X, Baudry M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells. 2020; 9(12):2698. https://doi.org/10.3390/cells9122698
Chicago/Turabian StyleWang, Yubin, Yan Liu, Xiaoning Bi, and Michel Baudry. 2020. "Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death" Cells 9, no. 12: 2698. https://doi.org/10.3390/cells9122698
APA StyleWang, Y., Liu, Y., Bi, X., & Baudry, M. (2020). Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells, 9(12), 2698. https://doi.org/10.3390/cells9122698