Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling

 , ,
, ,  , and
, and
Abstract
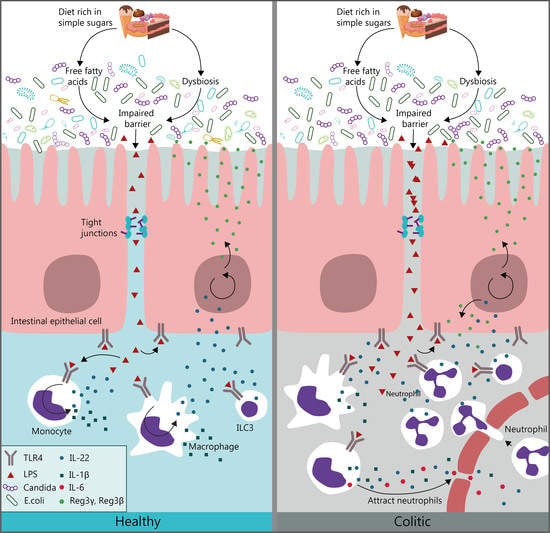
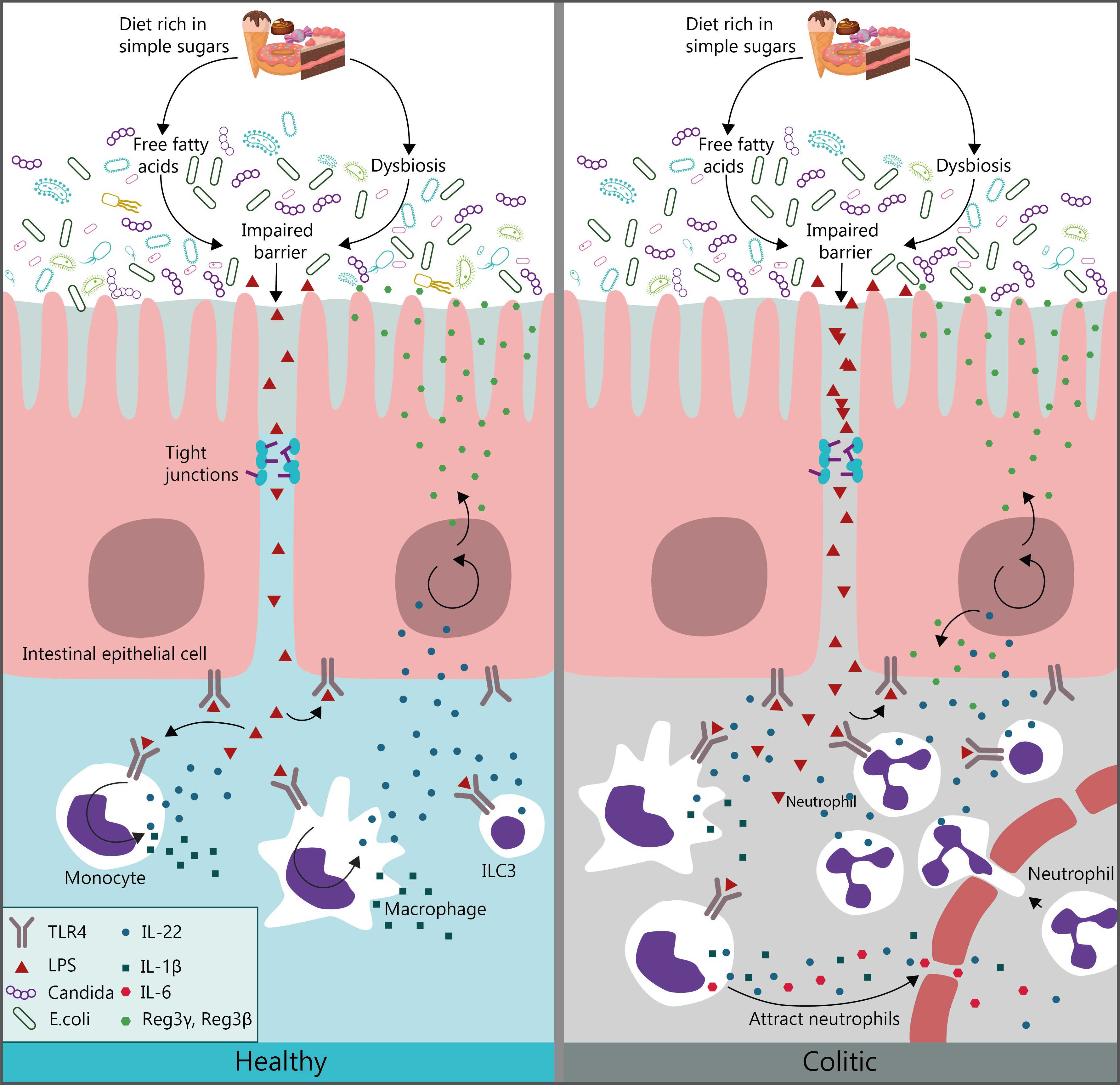
:
1. Introduction
2. Materials and Methods
2.1. Mice, Experimental Diets, and Gut Permeability
2.2. Experimental Mouse Models
2.3. Cell Suspension Preparation and Flow Cytometry
2.4. Cell Cultivation, Tissue Cultures, and Cytokine Measurement
2.5. Isolation of Total RNA, Reverse Transcription, and qPCR
2.6. Microbiome Analysis
2.7. Metabolome Analysis
2.8. Statistical Analysis
3. Results
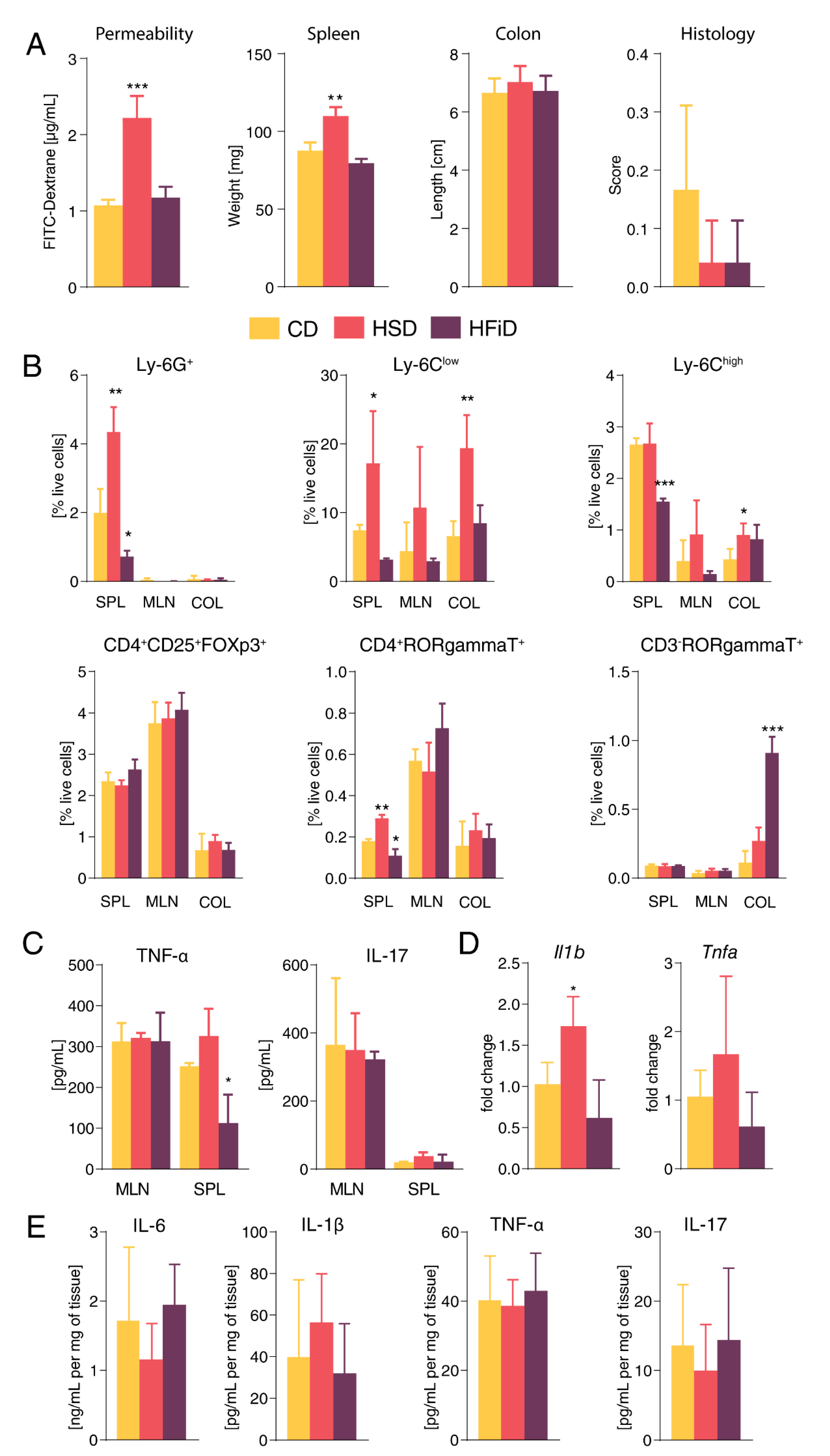
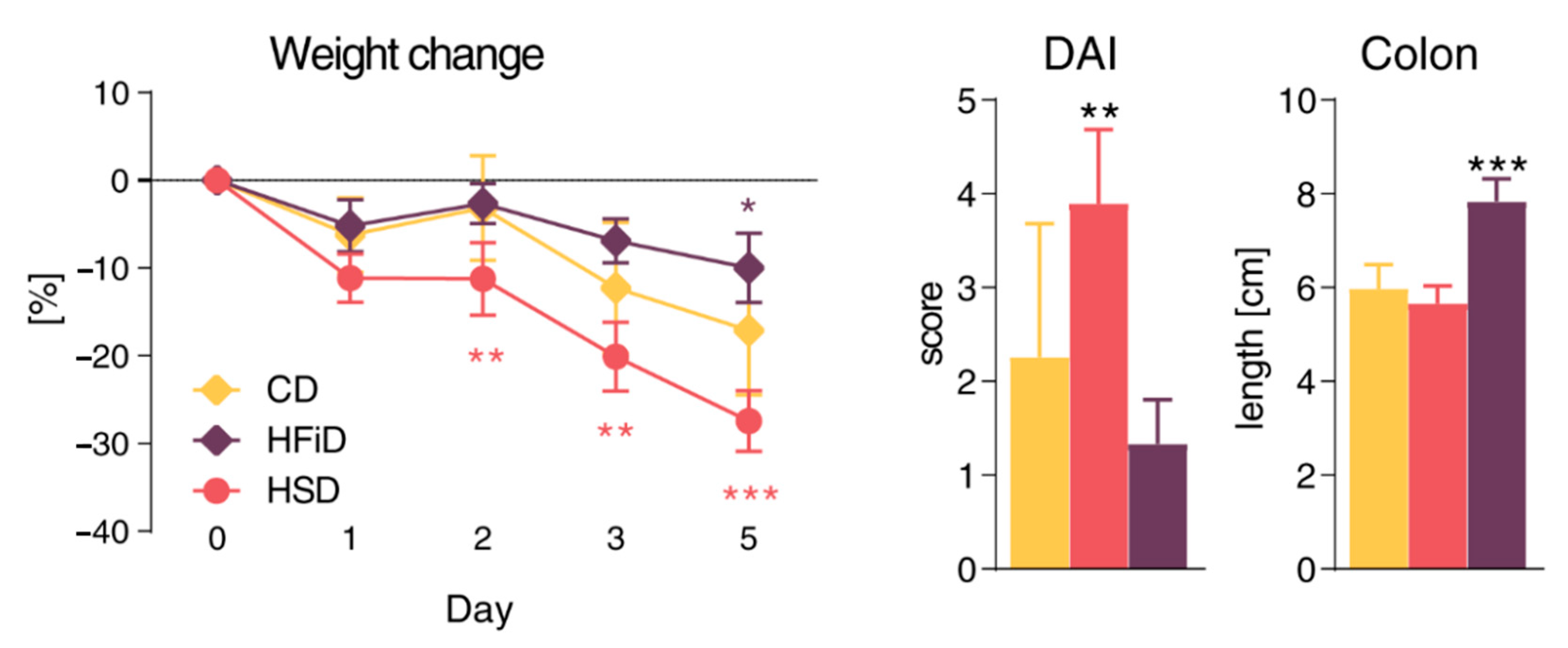
3.1. Diet Rich in Simple Carbohydrates Promotes Pro-Inflammatory Tuning
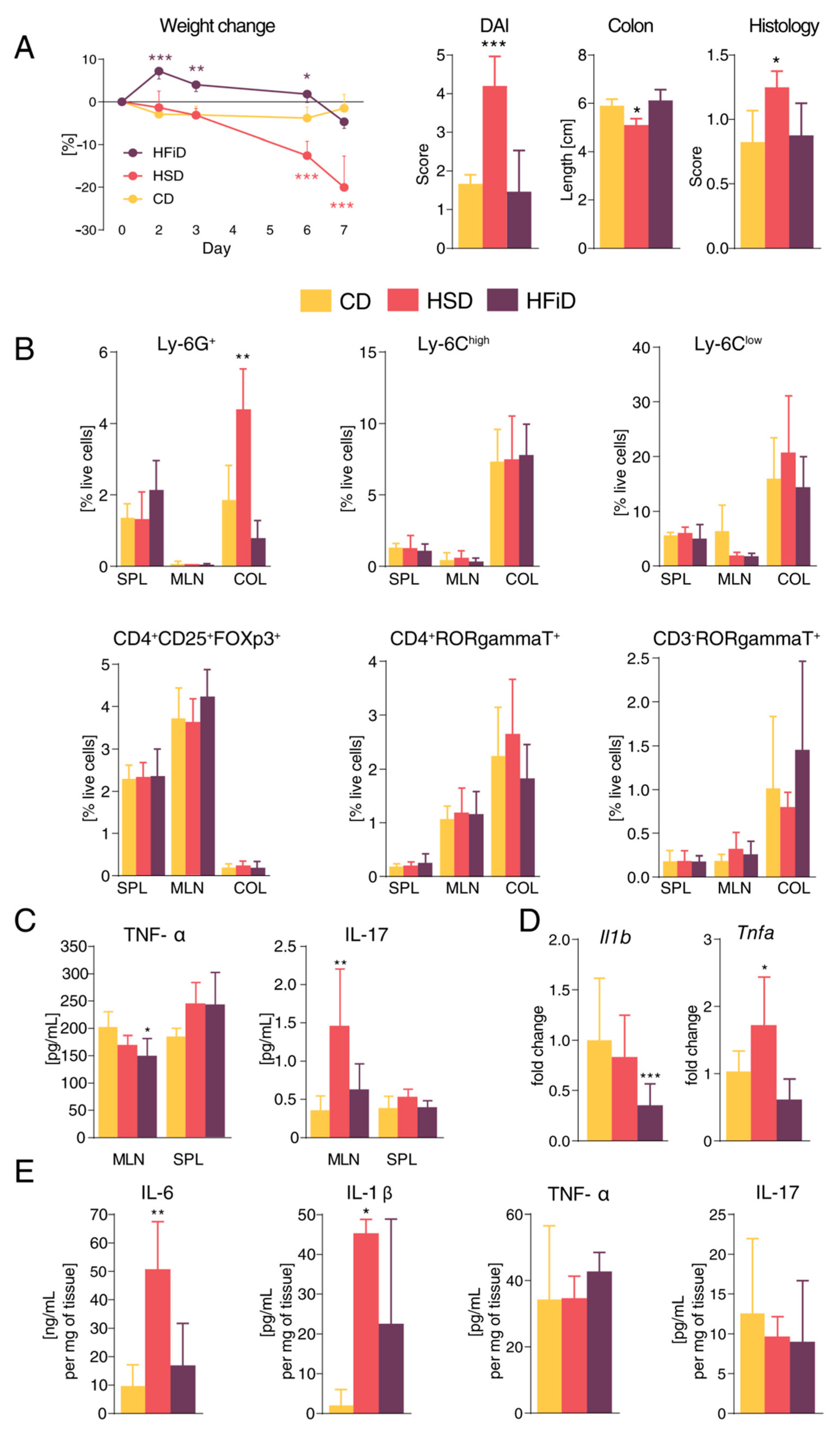
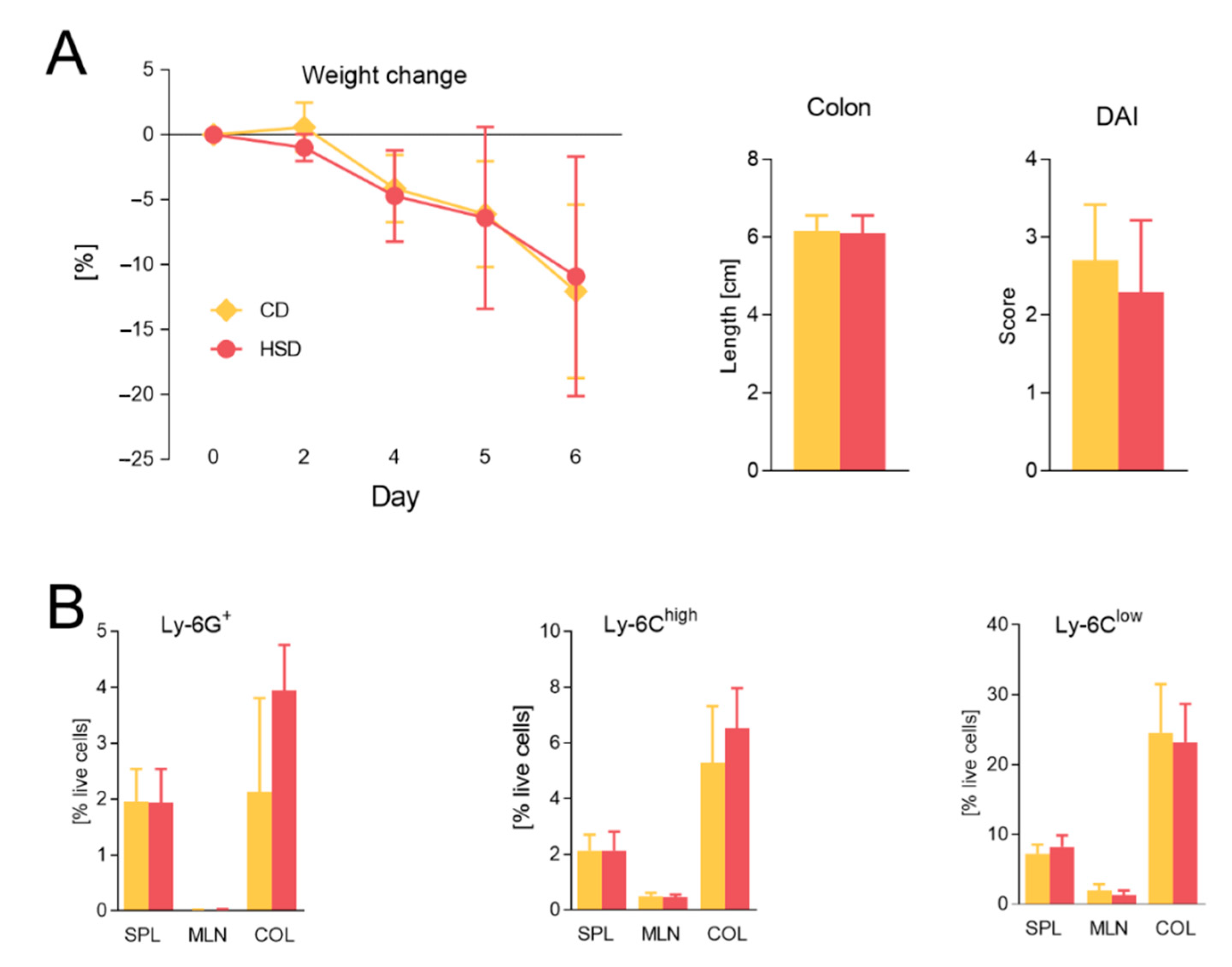
3.2. Diet Rich in Simple Sugars Worsens Acute DSS Colitis in a T-Cell-Independent Manner
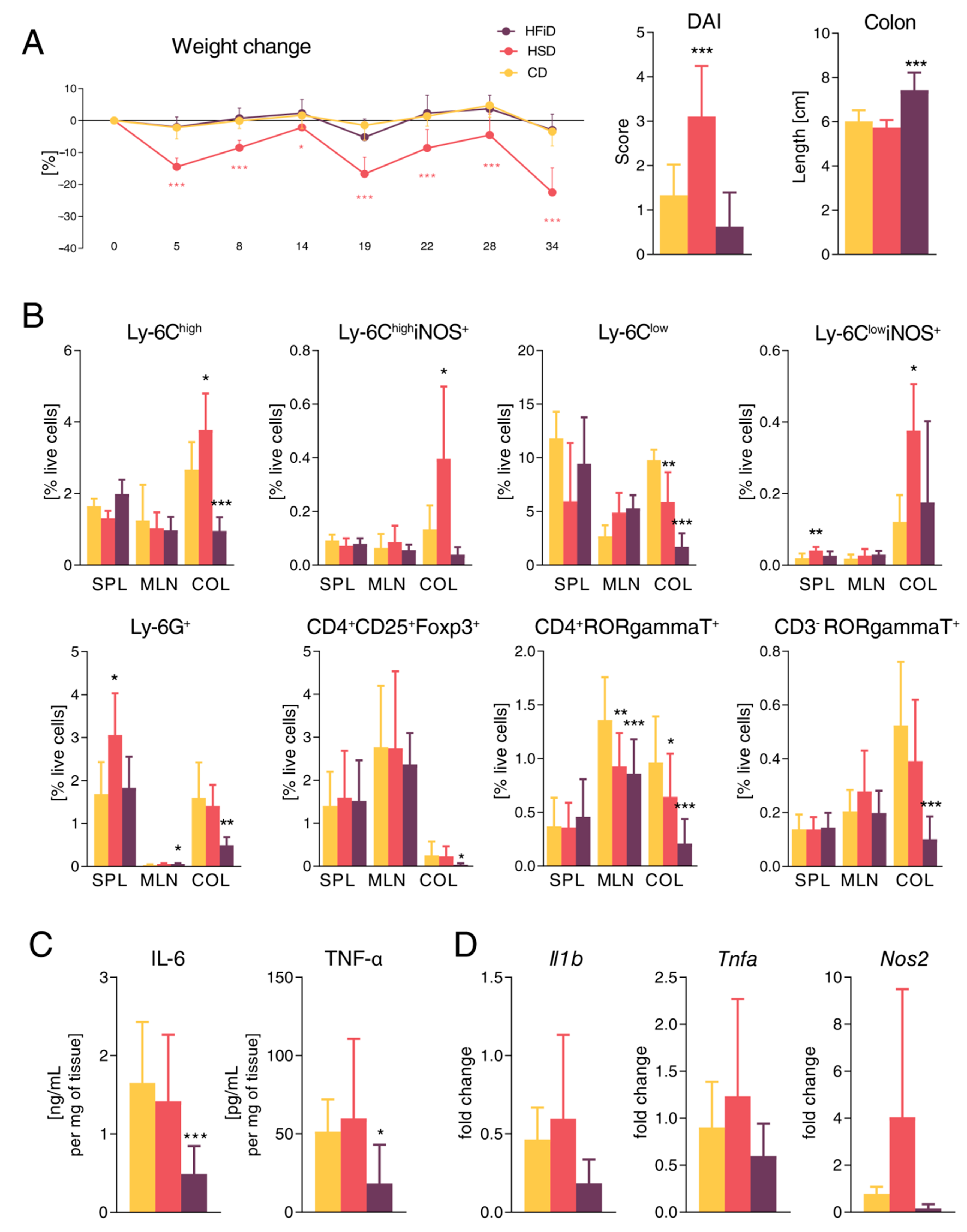
3.3. Diet Rich in Simple Carbohydrates Increases Severity of Chronic Colitis
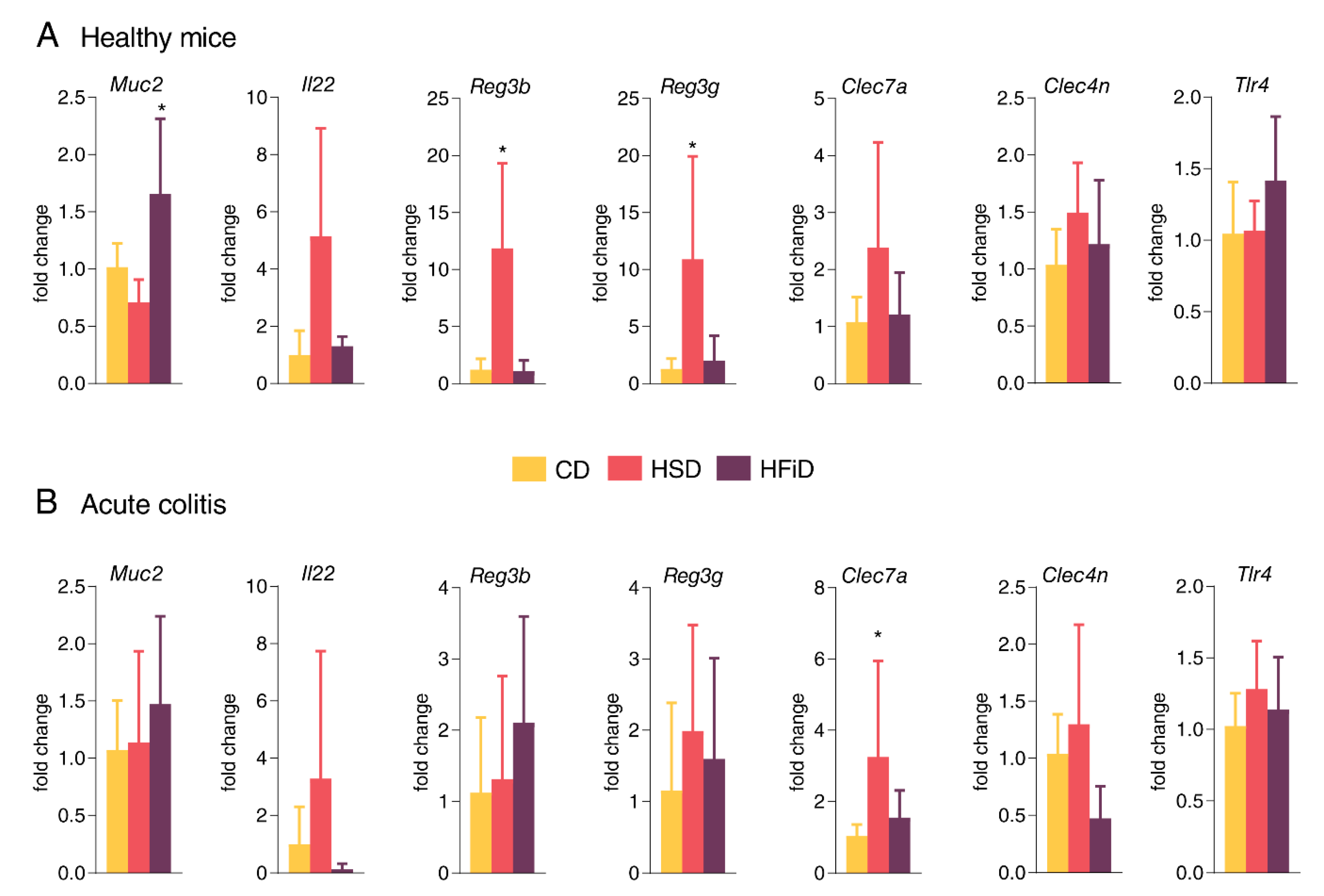
3.4. HSD Changes the Expression of Gut Barrier-Associated Genes in Colon
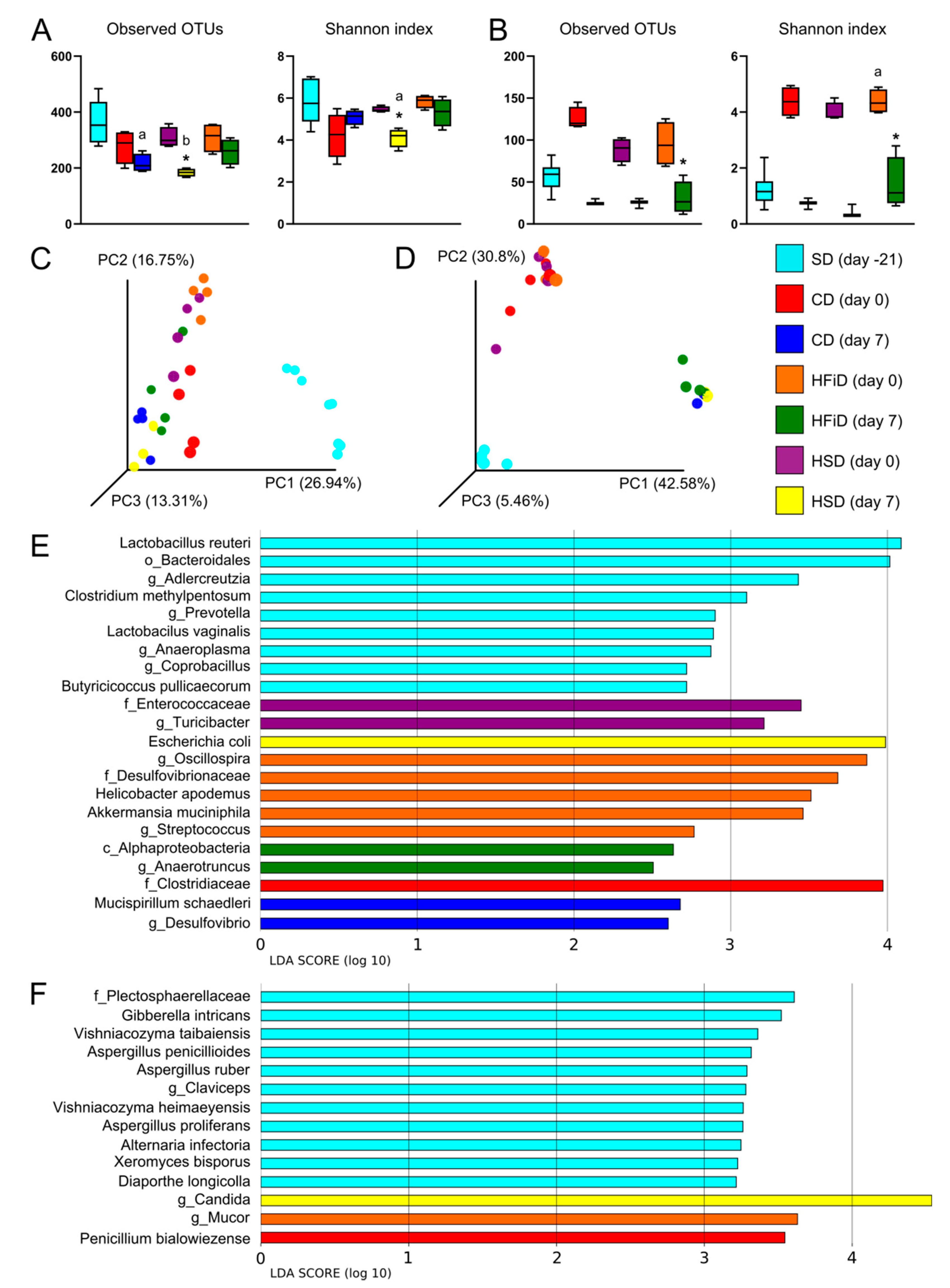
3.5. Diet Significantly Influenced Gut Bacterial and Fungal Microbiota Composition
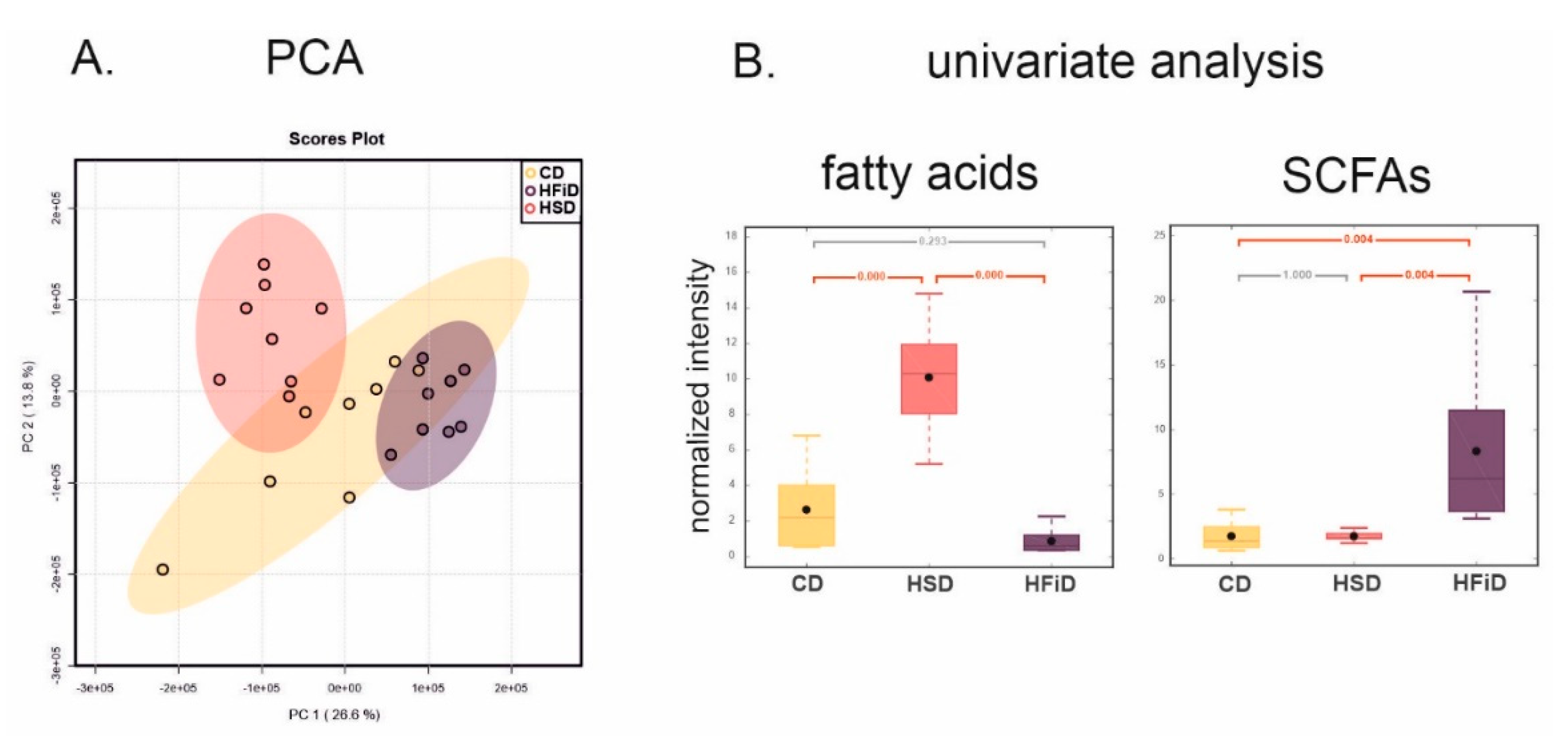
3.6. Diet Significantly Influences Fecal Metabolic Profile
3.7. HSD Detrimental Effect Is Not Transferred by Microbiota Alone
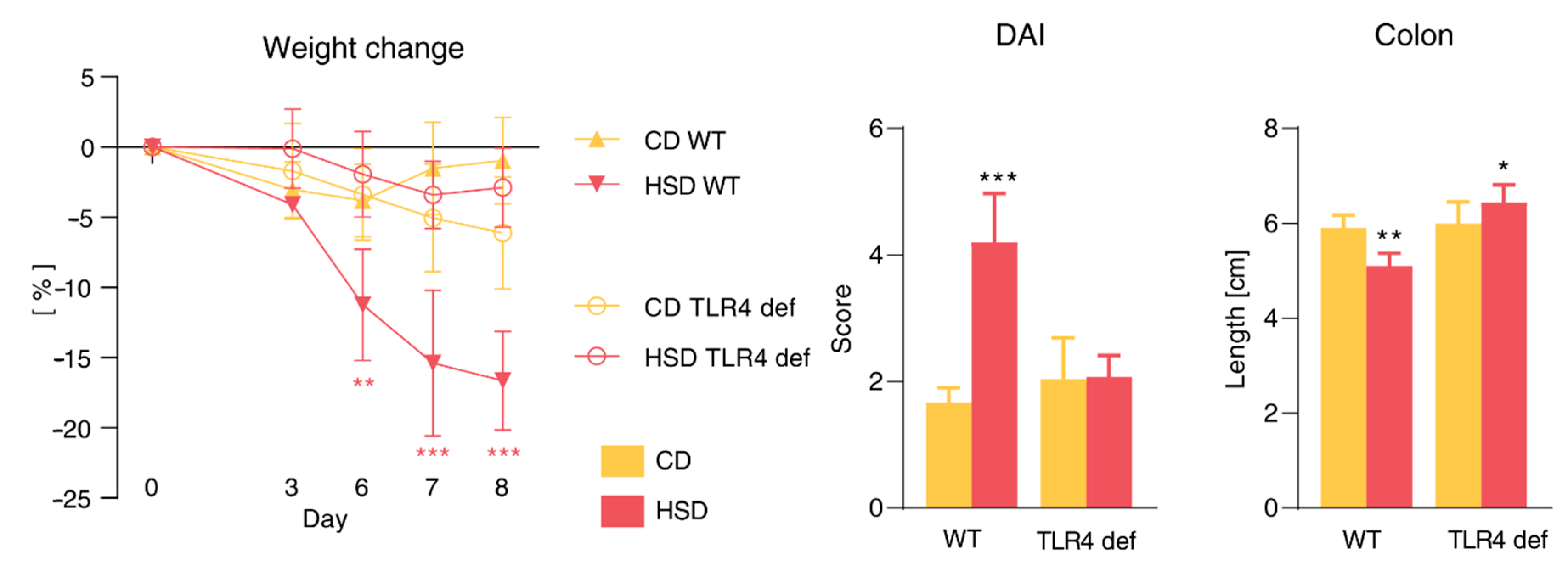
3.8. Pro-Inflammatory Effect of HSD Is Abrogated in TLR4-Deficient Mice and Is Not Limited to the Gut
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Filippo, C.; Lionetti, P. Impact of Diet on Gut Microbiota in the Globalized World. Funct. Food Rev. 2013, 5, 13–22. [Google Scholar] [CrossRef]
- Popkin, B.M.; Nielsen, S.J. The sweetening of the world’s diet. Obes. Res. 2003, 11, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, Z.; Gregg, E.W.; Flanders, W.D.; Merritt, R.; Hu, F.B. Added sugar intake and cardiovascular diseases mortality among us adults. JAMA Intern. Med. 2014, 174, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knüppel, A.; Shipley, M.J.; Llewellyn, C.H.; Brunner, E.J. Sugar intake from sweet food and beverages, common mental disorder and depression: Prospective findings from the Whitehall II study. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, B.C.M.; Wells, J.C.K.; Brayne, C.; Albanese, E.; Siervo, M. Increased fructose intake as a risk factor for dementia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65A, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Bédard, A.; Northstone, K.; Henderson, A.J.; Shaheen, S.O. Maternal intake of sugar during pregnancy and childhood respiratory and atopic outcomes. Eur. Respir. J. 2017, 50, 1700073. [Google Scholar] [CrossRef]
- Hwang, I.S.; Ho, H.; Hoffman, B.B.; Reaven, G.M. Fructose-induced insulin resistance and hypertension in rats. Hypertension 1987, 10, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.P.; Bartness, T.J.; Mielke, J.G.; Parent, M.B. A high fructose diet impairs spatial memory in male rats. Neurobiol. Learn. Mem. 2009, 92, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Alzoubi, K.H.; Khabour, O.F.; Salah, H.A.; Hasan, Z. Vitamin E prevents high-fat high-carbohydrates diet-induced memory impairment: The role of oxidative stress. Physiol. Behav. 2013, 119, 72–78. [Google Scholar] [CrossRef]
- Beilharz, J.E.; Maniam, J.; Morris, M.J. Short exposure to a diet rich in both fat and sugar or sugar alone impairs place, but not object recognition memory in rats. Brain. Behav. Immun. 2014, 37, 134–141. [Google Scholar] [CrossRef]
- Choudhary, P.; Pacholko, A.G.; Palaschuk, J.; Bekar, L.K. The locus coeruleus neurotoxin, DSP4, and/or a high sugar diet induce behavioral and biochemical alterations in wild-type mice consistent with Alzheimers related pathology. Metab. Brain Dis. 2018, 33, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Rock, E.; Gueux, E.; Mazur, A.; Grolier, P.; Rayssiguier, Y. Short-term consumption of a high-sucrose diet has a pro-oxidant effect in rats. Br. J. Nutr. 2002, 87, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Busserolles, J.; Zimowska, W.; Rock, E.; Rayssiguier, Y.; Mazur, A. Rats fed a high sucrose diet have altered heart antioxidant enzyme activity and gene expression. Life Sci. 2002, 71, 1303–1312. [Google Scholar] [CrossRef]
- Busserolles, J.; Gueux, E.; Rock, E.; Mazur, A.; Rayssiguier, Y. Substituting honey for refined carbohydrates protects rats from hypertriglyceridemic and prooxidative effects of fructose. J. Nutr. 2002, 132, 3379–3382. [Google Scholar] [CrossRef]
- Maslowski, K.M.; Mackay, C.R. Diet, gut microbiota and immune responses. Nat. Immunol. 2011, 12, 5–9. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Laffin, M.; Fedorak, R.; Zalasky, A.; Park, H.; Gill, A.; Agrawal, A.; Keshteli, A.; Hotte, N.; Madsen, K.L. A high-sugar diet rapidly enhances susceptibility to colitis via depletion of luminal short-chain fatty acids in mice. Sci. Rep. 2019, 9, 12294. [Google Scholar] [CrossRef] [Green Version]
- Do, M.H.; Lee, E.; Oh, M.J.; Kim, Y.; Park, H.Y. High-glucose or-fructose diet cause changes of the gut microbiota and metabolic disorders in mice without body weight change. Nutrients 2018, 10, 761. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic T reg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [Green Version]
- Tlaskalová-Hogenová, H.; Štěpánková, R.; Kozáková, H.; Hudcovic, T.; Vannucci, L.; Tučková, L.; Rossmann, P.; Hrnčíř, T.; Kverka, M.; Zákostelská, Z.; et al. The role of gut microbiota (commensal bacteria) and the mucosal barrier in the pathogenesis of inflammatory and autoimmune diseases and cancer: Contribution of germ-free and gnotobiotic animal models of human diseases. Cell. Mol. Immunol. 2011, 8, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, K.; Dudeck, A.; Hasenberg, M.; Nye, E.; Van Rooijen, N.; Hartmann, K.; Gunzer, M.; Roers, A.; Hogg, N. Mast cell and macrophage chemokines CXCL1/CXCL2 control the early stage of neutrophil recruitment during tissue inflammation. Blood 2013, 121, 4930–4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.I.; Ernst, R.K.; Bader, M.W. LPS, TLR4 and infectious disease diversity. Nat. Rev. Microbiol. 2005, 3, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [Green Version]
- Velloso, L.A.; Folli, F.; Saad, M.J. TLR4 at the crossroads of nutrients, gut microbiota, and metabolic inflammation. Endocr. Rev. 2015, 36, 245–271. [Google Scholar] [CrossRef] [Green Version]
- Chiba, M.; Nakane, K.; Komatsu, M. Westernized Diet is the Most Ubiquitous Environmental Factor in Inflammatory Bowel Disease. Perm. J. 2019, 23, 18–107. [Google Scholar] [CrossRef]
- Rizzello, F.; Spisni, E.; Giovanardi, E.; Imbesi, V.; Salice, M.; Alvisi, P.; Valerii, M.C.; Gionchetti, P. Implications of the westernized diet in the onset and progression of IBD. Nutrients 2019, 11, 1033. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive, E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.S.M.; Luben, R.; Van Schaik, F.; Oldenburg, B.; Bueno-De-Mesquita, H.B.; Hallmans, G.; Karling, P.; Lindgren, S.; Grip, O.; Key, T.; et al. Carbohydrate intake in the etiology of Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2014, 20, 2013–2021. [Google Scholar] [CrossRef] [Green Version]
- Racine, A.; Carbonnel, F.; Chan, S.S.M.; Hart, A.R.; Bueno-de-Mesquita, H.B.; Oldenburg, B.; van Schaik, F.D.M.; Tjønneland, A.; Olsen, A.; Dahm, C.C.; et al. Dietary Patterns and Risk of Inflammatory Bowel Disease in Europe. Inflamm. Bowel Dis. 2016, 22, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, K.; Kanmura, S.; Morinaga, Y.; Tanaka, A.; Makino, T.; Fujita, T.; Arima, S.; Sasaki, F.; Nasu, Y.; Tanoue, S.; et al. A high-fructose diet induces epithelial barrier dysfunction and exacerbates the severity of dextran sulfate sodium-induced colitis. Int. J. Mol. Med. 2019, 43, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Montrose, D.C.; Nishiguchi, R.; Basu, S.; Staab, H.A.; Zhou, X.K.; Wang, H.; Meng, L.; Johncilla, M.; Cubillos-Ruiz, J.R.; Morales, D.K.; et al. Dietary Fructose Alters the Composition, Localization and Metabolism of Gut Microbiota in Association with Worsening Colitis. Cell. Mol. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Waliullah, S.; Godfrey, V.; Khan, M.A.W.; Ramachandran, R.A.; Cantarel, B.L.; Behrendt, C.; Peng, L.; Hooper, L.V.; Zaki, H. Dietary simple sugars alter microbial ecology in the gut and promote colitis in mice. Sci. Transl. Med. 2020, 12, eaay6218. [Google Scholar] [CrossRef] [PubMed]
- Zakostelska, Z.; Kverka, M.; Klimesova, K.; Rossmann, P.; Mrazek, J.; Kopecny, J.; Hornova, M.; Srutkova, D.; Hudcovic, T.; Ridl, J.; et al. Lysate of probiotic Lactobacillus casei DN-114 001 ameliorates colitis by strengthening the gut barrier function and changing the gut microenvironment. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Kostovcikova, K.; Coufal, S.; Galanova, N.; Fajstova, A.; Hudcovic, T.; Kostovcik, M.; Prochazkova, P.; Zakostelska, Z.J.; Cermakova, M.; Sediva, B.; et al. Diet rich in animal protein promotes pro-inflammatory macrophage response and exacerbates colitis in mice. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Couter, C.J.; Surana, N.K. Isolation and flow cytometric characterization of murine small intestinal lymphocytes. J. Vis. Exp. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Findley, K.; Oh, J.; Yang, J.; Conlan, S.; Deming, C.; Meyer, J.A.; Schoenfeld, D.; Nomicos, E.; Park, M.; Becker, J.; et al. Topographic diversity of fungal and bacterial communities in human skin. Nature 2013, 498, 367–370. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pẽa, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Jacob, D.; Deborde, C.; Lefebvre, M.; Maucourt, M.; Moing, A. NMRProcFlow: A graphical and interactive tool dedicated to 1D spectra processing for NMR-based metabolomics. Metabolomics 2017, 13, 36. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68. [Google Scholar] [CrossRef]
- Dieterle, F.; Ross, A.; Schlotterbeck, G.; Senn, H. Probabilistic quotient normalization as robust method to account for dilution of complex biological mixtures. Application in1H NMR metabonomics. Anal. Chem. 2006, 78, 4281–4290. [Google Scholar] [CrossRef]
- Manzel, A.; Muller, D.N.; Hafler, D.A.; Erdman, S.E.; Linker, R.A.; Kleinewietfeld, M. Role of “western diet” in inflammatory autoimmune diseases. Curr. Allergy Asthma Rep. 2014, 14, 404. [Google Scholar] [CrossRef] [Green Version]
- Key, T.J.; Allen, N.E.; Spencer, E.A.; Travis, R.C. The effect of diet on risk of cancer. Lancet 2002, 360, 861–868. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Leon Coria, A.; Cornick, S.; Petri, B.; Mayengbam, S.; Jijon, H.B.; Moreau, F.; Shearer, J.; Chadee, K. Increased intestinal permeability exacerbates sepsis through reduced hepatic SCD-1 activity and dysregulated iron recycling. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kawada, M.; Arihiro, A.; Mizoguchi, E. Insights from advances in research of chemically induced experimental models of human inflammatory bowel disease. World J. Gastroenterol. 2007, 13, 5581–5593. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.; Heinbockel, L.; Brandenburg, K.; Hornef, M.W. Antimicrobial peptides and the enteric mucus layer act in concert to protect the intestinal mucosa. Gut Microbes 2015, 5, 761–765. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, A.; Yano, A.; Himuro, H.; Ezaki, Y.; Sadanaga, T.; Mizoguchi, E. Clinical importance of IL-22 cascade in IBD. J. Gastroenterol. 2018, 53, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [Green Version]
- Aden, K.; Rehman, A.; Falk-Paulsen, M.; Secher, T.; Kuiper, J.; Tran, F.; Pfeuffer, S.; Sheibani-Tezerji, R.; Breuer, A.; Luzius, A.; et al. Epithelial IL-23R Signaling Licenses Protective IL-22 Responses in Intestinal Inflammation. Cell Rep. 2016, 16, 2208–2218. [Google Scholar] [CrossRef] [Green Version]
- Zindl, C.L.; Lai, J.F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 12768–12773. [Google Scholar] [CrossRef] [Green Version]
- Keir, M.E.; Yi, T.; Lu, T.T.; Ghilardi, N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Pelczar, P.; Witkowski, M.; Perez, L.G.; Kempski, J.; Hammel, A.G.; Brockmann, L.; Kleinschmidt, D.; Wende, S.; Haueis, C.; Bedke, T.; et al. A pathogenic role for T cell-derived IL-22BP in inflammatory bowel disease. Science 2016, 354, 358–362. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Di Paola, M.; Ramazzotti, M.; Albanese, D.; Pieraccini, G.; Banci, E.; Miglietta, F.; Cavalieri, D.; Lionetti, P. Diet, Environments, and Gut Microbiota. A Preliminary Investigation in Children Living in Rural and Urban Burkina Faso and Italy. Front. Microbiol. 2017, 8, 1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konikoff, T.; Gophna, U. Oscillospira: A Central, Enigmatic Component of the Human Gut Microbiota. Trends Microbiol. 2016, 24, 523–524. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; de Vos, W.M. Next-generation beneficial microbes: The case of Akkermansia muciniphila. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Seregin, S.S.; Golovchenko, N.; Schaf, B.; Chen, J.; Pudlo, N.A.; Mitchell, J.; Baxter, N.T.; Zhao, L.; Schloss, P.D.; Martens, E.C.; et al. NLRP6 Protects Il10−/− Mice from Colitis by Limiting Colonization of Akkermansia muciniphila. Cell Rep. 2017, 19, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef]
- Bertolini, M.; Ranjan, A.; Thompson, A.; Diaz, P.I.; Sobue, T.; Maas, K.; Dongari-Bagtzoglou, A. Candida albicans induces mucosal bacterial dysbiosis that promotes invasive infection. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef]
- Boudeau, J.; Glasser, A.L.; Masseret, E.; Joly, B.; Darfeuille-Michaud, A. Invasive ability of an Escherichia coli strain isolated from the ileal mucosa of a patient with Crohn’s disease. Infect. Immun. 1999, 67, 4499–4509. [Google Scholar] [CrossRef] [Green Version]
- Panpetch, W.; Hiengrach, P.; Nilgate, S.; Tumwasorn, S.; Somboonna, N.; Wilantho, A.; Chatthanathon, P.; Prueksapanich, P.; Leelahavanichkul, A. Additional Candida albicans administration enhances the severity of dextran sulfate solution induced colitis mouse model through leaky gut-enhanced systemic inflammation and gut-dysbiosis but attenuated by Lactobacillus rhamnosus L34. Gut Microbes 2020, 11, 465–480. [Google Scholar] [CrossRef]
- Sovran, B.; Planchais, J.; Jegou, S.; Straube, M.; Lamas, B.; Natividad, J.M.; Agus, A.; Dupraz, L.; Glodt, J.; Da Costa, G.; et al. Enterobacteriaceae are essential for the modulation of colitis severity by fungi. Microbiome 2018, 6, 152. [Google Scholar] [CrossRef]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, Colonic Fermentation, and Gastrointestinal Health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Louis, P.; Flint, H.J. Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol. 2004, 70, 5810–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerrum, J.T.; Wang, Y.; Hao, F.; Coskun, M.; Ludwig, C.; Günther, U.; Nielsen, O.H. Metabonomics of human fecal extracts characterize ulcerative colitis, Crohn’s disease and healthy individuals. Metabolomics 2014, 11, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernia, P.; Caprilli, R.; Latella, G.; Barbetti, F.; Magliocca, F.M.; Cittadini, M. Fecal Lactate and Ulcerative Colitis. Gastroenterology 1988, 95, 1564–1568. [Google Scholar] [CrossRef]
- Hashizume, K.; Tsukahara, T.; Yamada, K.; Koyama, H.; Ushida, K. Megasphaera elsdenii JCM1772T normalizes hyperlactate production in the large intestine of fructooligosaccharide-fed rats by stimulating butyrate production. J. Nutr. 2003, 133, 3187–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.; Macfarlane, G. Enumeration of amino acid fermenting bacteria in the human large intestine: Effects of pH and starch on peptide metabolism and dissimilation of amino acids. FEMS Microbiol. Ecol. 1998, 25, 355–368. [Google Scholar] [CrossRef]
- Wang, M.; Xie, Z.; Li, L.; Chen, Y.; Li, Y.; Wang, Y.; Lu, B.; Zhang, S.; Ma, F.; Ma, C.W.; et al. Supplementation with compound polysaccharides contributes to the development and metabolic activity of young rat intestinal microbiota. Food Funct. 2019, 10, 2658–2675. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hernandez, L.D.; Galán, J.E.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Biswas, A.; Wilmanski, J.; Forsman, H.; Hrncir, T.; Hao, L.; Tlaskalova-Hogenova, H.; Kobayashi, K.S. Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur. J. Immunol. 2011, 41, 182–194. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HFiD vs. CD | HSD vs. CD | HFiD vs. HSD | ||||
|---|---|---|---|---|---|---|
| Δ (%) | p-Value | ∆ (%) | p-Value | ∆ (%) | p-Value | |
| SCFAs | ||||||
| Acetate | 369.74 | 0.0056 | −1.83 | 0.9998 | 378.48 | 0.0054 |
| Propionate | 461.72 | 0.0002 | 13.85 | 0.9885 | 393.38 | 0.0004 |
| Amino acids | ||||||
| Alanine | −31.70 | 0.2688 | 47.74 | 0.0631 | −53.77 | 0.0018 |
| Aspartate | −51.09 | 0.0107 | 22.51 | 0.3469 | −60.08 | 0.0004 |
| Glutamate | −35.44 | 0.1888 | 34.15 | 0.2108 | −51.88 | 0.0050 |
| Isoleucine | −22.23 | 0.6574 | 46.09 | 0.1849 | −46.77 | 0.0337 |
| Lysine | −50.35 | 0.0485 | 14.14 | 0.7591 | −56.50 | 0.0103 |
| Methionine | −43.22 | 0.0935 | 55.89 | 0.0248 | −63.58 | 0.0001 |
| Phenylalanine | −26.49 | 0.3116 | 40.13 | 0.0824 | −47.54 | 0.0031 |
| Tyrosine | −55.35 | 0.0191 | 30.80 | 0.2460 | −65.86 | 0.0004 |
| Others | ||||||
| Glucose | −66.04 | 0.0000 | −48.78 | 0.0006 | −33.70 | 0.2779 |
| Lactate | −40.35 | 0.0074 | −9.82 | 0.6917 | −33.85 | 0.0455 |
| Fumarate | 47.72 | 0.0195 | 9.66 | 0.8221 | 34.72 | 0.0688 |
| Bile acids a | −67.97 | 0.0206 | −34.37 | 0.3179 | −51.19 | 0.3336 |
| Methanol | 4.45 | 0.7509 | −16.25 | 0.0383 | 24.71 | 0.0077 |
| Uracil | −62.30 | 0.0043 | 16.47 | 0.6092 | −67.63 | 0.0004 |
| Fatty acids | −67.10 | 0.2928 | 283.60 | 0.0000 | −91.42 | 0.0000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fajstova, A.; Galanova, N.; Coufal, S.; Malkova, J.; Kostovcik, M.; Cermakova, M.; Pelantova, H.; Kuzma, M.; Sediva, B.; Hudcovic, T.; et al. Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells 2020, 9, 2701. https://doi.org/10.3390/cells9122701
Fajstova A, Galanova N, Coufal S, Malkova J, Kostovcik M, Cermakova M, Pelantova H, Kuzma M, Sediva B, Hudcovic T, et al. Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells. 2020; 9(12):2701. https://doi.org/10.3390/cells9122701
Chicago/Turabian StyleFajstova, Alena, Natalie Galanova, Stepan Coufal, Jana Malkova, Martin Kostovcik, Martina Cermakova, Helena Pelantova, Marek Kuzma, Blanka Sediva, Tomas Hudcovic, and et al. 2020. "Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling" Cells 9, no. 12: 2701. https://doi.org/10.3390/cells9122701
APA StyleFajstova, A., Galanova, N., Coufal, S., Malkova, J., Kostovcik, M., Cermakova, M., Pelantova, H., Kuzma, M., Sediva, B., Hudcovic, T., Hrncir, T., Tlaskalova-Hogenova, H., Kverka, M., & Kostovcikova, K. (2020). Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells, 9(12), 2701. https://doi.org/10.3390/cells9122701