Targeting Aggrephagy for the Treatment of Alzheimer’s Disease

 , , and
, , and
Abstract
:1. Introduction
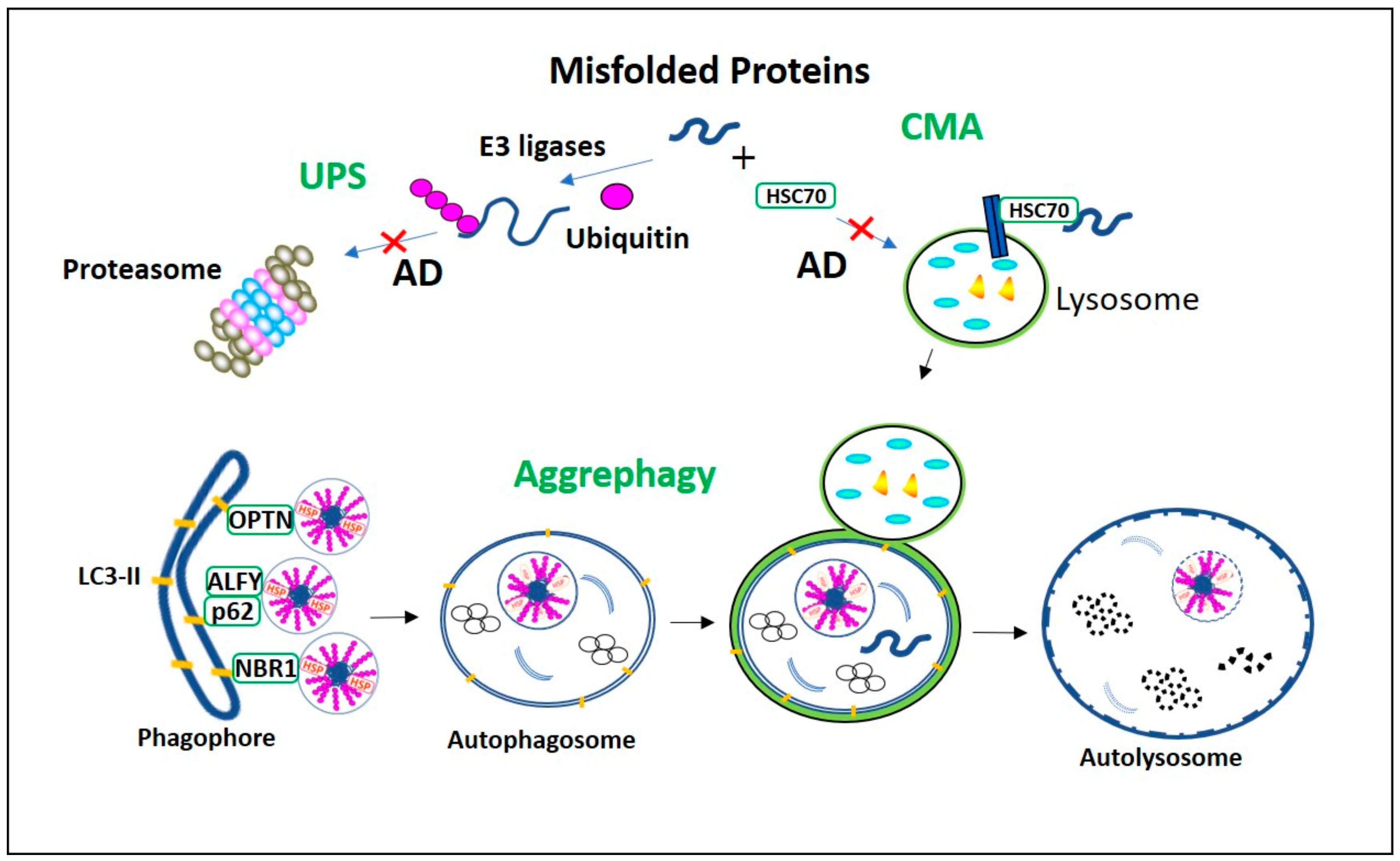
2. Process and Regulation of Aggrephagy
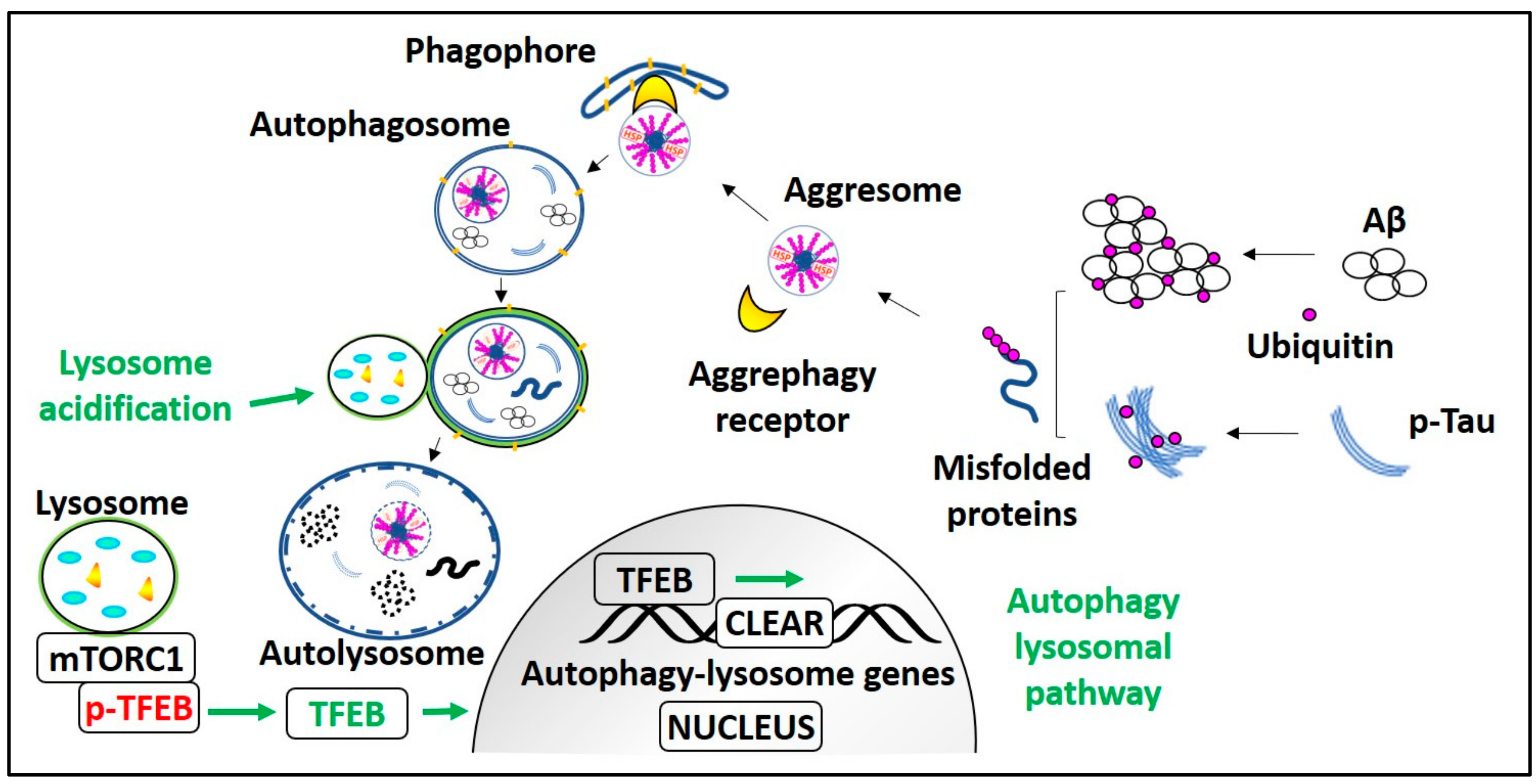
2.1. Aggresome Formation
2.2. Aggrephagy Regulation
2.3. Role of Aggrephagy Receptors
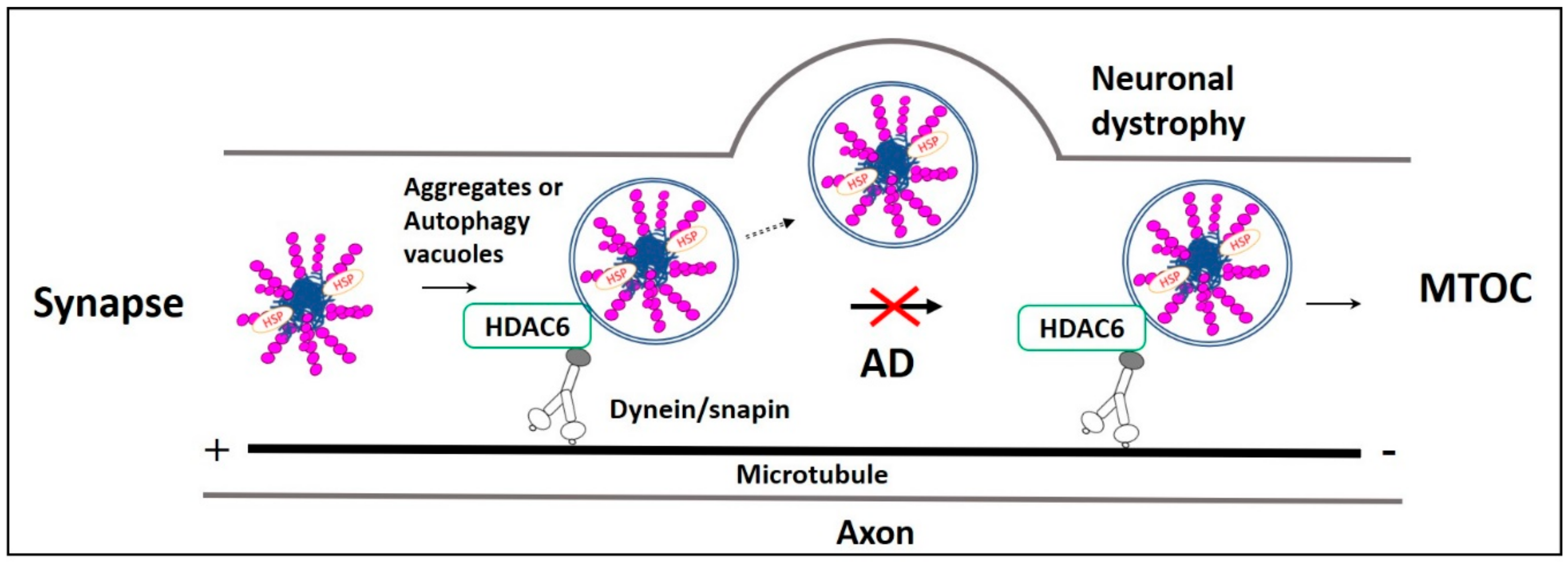
2.4. Role of HDAC6 in Aggrephagy
3. Aggrephagy Dysfunction in AD
3.1. Aggresomes in AD
3.2. Dysregulation in Aggregates Retrograde Transport in AD
3.3. Aggrephagy Receptor Impairment
4. Aggrephagy as a Therapeutic Target for the Treatment of AD
4.1. Genetic Approach
4.2. Lysosomes Functional Restoration with Nanotechnology
4.3. Small Molecule Aggrephagy Enhancers on AD Models
4.3.1. mTOR-Inhibiting Aggrephagy Inducers
4.3.2. TFEB-Activating Aggrephagy Inducers
5. Conclusions and Future Direction
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kelley, B.J.; Petersen, R.C. Alzheimer’s disease and mild cognitive impairment. Neurol. Clin. 2007, 25, 577–609. [Google Scholar] [CrossRef] [Green Version]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [Green Version]
- Schindowski, K.; Bretteville, A.; Leroy, K.; Bégard, S.; Brion, J.P.; Hamdane, M.; Buée, L. Alzheimer’s Disease-Like Tau Neuropathology Leads to Memory Deficits and Loss of Functional Synapses in a Novel Mutated Tau Transgenic Mouse without Any Motor Deficits. Am. J. Pathol. 2006, 169, 599–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lyubchenko, Y.L. The structure of misfolded amyloidogenic dimers: Computational analysis of force spectroscopy data. Biophys. J. 2014, 107, 2903–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassar, R. BACE1: The beta-secretase enzyme in Alzheimer’s disease. J. Mol. Neurosci. 2004, 23, 105–114. [Google Scholar] [CrossRef]
- Iwatsubo, T. The gamma-secretase complex: Machinery for intramembrane proteolysis. Curr. Opin. Neurobiol. 2004, 14, 379–383. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prevot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y.W. The gamma-secretase complex: From structure to function. Front. Cell Neurosci. 2015, 8, 427. [Google Scholar] [CrossRef] [Green Version]
- Ricciarelli, R.; Fedele, E. The Amyloid Cascade Hypothesis in Alzheimer’s Disease: It’s Time to Change Our Mind. Curr. Neuropharmacol. 2017, 15, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Perl, D.P. Neuropathology of Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Haque, M.M.; Kim, D.; Kim, D.J.; Kim, Y.K. Cell-based Models to Investigate Tau Aggregation. Comput. Struct. Biotechnol. J. 2014, 12, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleary, J.P.; Walsh, D.M.; Hofmeister, J.J.; Shankar, G.M.; Kuskowski, M.A.; Selkoe, D.J.; Ashe, K.H. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat. Neurosci. 2005, 8, 79–84. [Google Scholar] [CrossRef]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A. Intracellular protein degradation: From a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Best Pract. Res. Clin. Haematol. 2017, 30, 341–355. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo Recognition and Degradation by Selective Autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Koga, H.; Cuervo, A.M. Chaperone-mediated autophagy dysfunction in the pathogenesis of neurodegeneration. Neurobiol. Dis. 2011, 43, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Overbye, A.; Fengsrud, M.; Seglen, P.O. Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 2007, 3, 300–322. [Google Scholar] [CrossRef] [Green Version]
- Barber, K.W.; Rinehart, J. The ABCs of PTMs. Nat. Chem. Biol. 2018, 14, 188–192. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Basso, M.; Samengo, G.; Nardo, G.; Massignan, T.; D’Alessandro, G.; Tartari, S.; Cantoni, L.; Marino, M.; Cheroni, C.; De Biasi, S.; et al. Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PLoS ONE 2009, 4, e8130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, W.H.; Choi, Y.P. In vitro generation of tau aggregates conformationally distinct from parent tau seeds of Alzheimer’s brain. Prion 2018, 13, 1–12. [Google Scholar] [CrossRef]
- De, S.; Wirthensohn, D.C.; Flagmeier, P.; Hughes, C.; Aprile, F.A.; Ruggeri, F.S.; Whiten, D.R.; Emin, D.; Xia, Z.; Varela, J.A.; et al. Different soluble aggregates of Abeta42 can give rise to cellular toxicity through different mechanisms. Nat. Commun. 2019, 10, 1541. [Google Scholar] [CrossRef] [Green Version]
- Fusco, G.; Chen, S.W.; Williamson, P.T.F.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N.; et al. Structural basis of membrane disruption and cellular toxicity by alpha-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Santa-Maria, I.; Varghese, M.; Ksiezak-Reding, H.; Dzhun, A.; Wang, J.; Pasinetti, G.M. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J. Biol. Chem. 2012, 287, 20522–20533. [Google Scholar] [CrossRef] [Green Version]
- Nassar, M.; Samaha, H.; Ghabriel, M.; Yehia, M.; Taha, H.; Salem, S.; Shaaban, K.; Omar, M.; Ahmed, N.; El-Naggar, S. LC3A Silencing Hinders Aggresome Vimentin Cage Clearance in Primary Choroid Plexus Carcinoma. Sci. Rep. 2017, 7, 8022. [Google Scholar] [CrossRef] [Green Version]
- Bardag-Gorce, F.; Makalinao, A.; Meepe, I.; Hoft, R.H.; Cortez, D.; Oliva, J.; Laporte, A.; Stark, J.; Gorce, A.; Di Lorenzo, M.; et al. Corneal keratin aggresome (CKAGG) formation and clearance by proteasome activation. Heliyon 2018, 4, e01012. [Google Scholar] [CrossRef]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Chigurupati, S.; Raymick, J.; Mann, D.; Bowyer, J.F.; Schmitt, T.; Beger, R.D.; Hanig, J.P.; Schmued, L.C.; Paule, M.G. Neuroprotective effect of the chemical chaperone, trehalose in a chronic MPTP-induced Parkinson’s disease mouse model. Neurotoxicology 2014, 44, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Mogk, A.; Bukau, B. Role of sHsps in organizing cytosolic protein aggregation and disaggregation. Cell Stress Chaperones 2017, 22, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Ungelenk, S.; Moayed, F.; Ho, C.T.; Grousl, T.; Scharf, A.; Mashaghi, A.; Tans, S.; Mayer, M.P.; Mogk, A.; Bukau, B. Small heat shock proteins sequester misfolding proteins in near-native conformation for cellular protection and efficient refolding. Nat. Commun. 2016, 7, 13673. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I. Ubiquitin signaling and autophagy. J. Biol. Chem. 2017, 293, 5404–5413. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol. 2003, 21, 921–926. [Google Scholar] [CrossRef]
- Ikeda, F.; Dikic, I. Atypical ubiquitin chains: New molecular signals. ’Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008, 9, 536–542. [Google Scholar] [CrossRef] [Green Version]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lystad, A.H.; Ichimura, Y.; Takagi, K.; Yang, Y.; Pankiv, S.; Kanegae, Y.; Kageyama, S.; Suzuki, M.; Saito, I.; Mizushima, T.; et al. Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep. 2014, 15, 557–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isakson, P.; Holland, P.; Simonsen, A. The role of ALFY in selective autophagy. Cell Death Differ. 2013, 20, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filimonenko, M.; Isakson, P.; Finley, K.D.; Anderson, M.; Jeong, H.; Melia, T.J.; Bartlett, B.J.; Myers, K.M.; Birkeland, H.C.; Lamark, T.; et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell. 2010, 38, 265–279. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Johansen, T.; Dikic, I. NBR1 cooperates with p62 in selective autophagy of ubiquitinated targets. Autophagy 2009, 5, 732–733. [Google Scholar] [CrossRef] [Green Version]
- Fusco, C.; Micale, L.; Egorov, M.; Monti, M.; D’Addetta, E.V.; Augello, B.; Cozzolino, F.; Calcagni, A.; Fontana, A.; Polishchuk, R.S.; et al. The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS ONE 2012, 7, e40440. [Google Scholar] [CrossRef] [Green Version]
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Shin, D.; Kwon, S.H. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2012, 280, 775–793. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Watabe, M.; Nakaki, T. Protein kinase CK2 regulates the formation and clearance of aggresomes in response to stress. J. Cell Sci. 2011, 124, 1519–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Li, L.; Chudaev, M.V.; Chen, J.; Perez, F.A.; Palmiter, R.D.; Chin, L.S. Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 2007, 178, 1025–1038. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, H.; Ali, Y.O.; Ravichandran, M.; Dong, A.; Qiu, W.; MacKenzie, F.; Dhe-Paganon, S.; Arrowsmith, C.H.; Zhai, R.G. Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 2011, 287, 2317–2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.P.; Zhou, D.; Ouyang, D.Y.; Xu, L.H.; Wang, Y.; Wang, L.X.; Pan, H.; He, X.H. LC3B-II deacetylation by histone deacetylase 6 is involved in serum-starvation-induced autophagic degradation. Biochem. Biophys. Res. Commun. 2013, 441, 970–975. [Google Scholar] [CrossRef]
- Lu, M.; Williamson, N.; Mishra, A.; Michel, C.H.; Kaminski, C.F.; Tunnacliffe, A.; Kaminski Schierle, G.S. Structural progression of amyloid-beta Arctic mutant aggregation in cells revealed by multiparametric imaging. J. Biol. Chem. 2018, 294, 1478–1487. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.; Huang, H.C.; Jiang, Z.F. Relationship between amyloid-beta and the ubiquitin-proteasome system in Alzheimer’s disease. Neurol. Res. 2014, 36, 276–282. [Google Scholar] [CrossRef]
- Morimoto, D.; Walinda, E.; Fukada, H.; Sou, Y.S.; Kageyama, S.; Hoshino, M.; Fujii, T.; Tsuchiya, H.; Saeki, Y.; Arita, K.; et al. The unexpected role of polyubiquitin chains in the formation of fibrillar aggregates. Nat. Commun. 2015, 6, 6116. [Google Scholar] [CrossRef] [Green Version]
- Lakshmana, M.K.; Chung, J.Y.; Wickramarachchi, S.; Tak, E.; Bianchi, E.; Koo, E.H.; Kang, D.E. A fragment of the scaffolding protein RanBP9 is increased in Alzheimer’s disease brains and strongly potentiates amyloid-beta peptide generation. FASEB J. 2009, 24, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Lakshmana, M.K.; Yoon, I.S.; Chen, E.; Bianchi, E.; Koo, E.H.; Kang, D.E. Novel role of RanBP9 in BACE1 processing of amyloid precursor protein and amyloid beta peptide generation. J. Biol. Chem. 2009, 284, 11863–11872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salemi, L.M.; Almawi, A.W.; Lefebvre, K.J.; Schild-Poulter, C. Aggresome formation is regulated by RanBPM through an interaction with HDAC6. Biol. Open 2014, 3, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, K.; Alonso, A.C.; Gong, C.X.; Khatoon, S.; Pei, J.J.; Wang, J.Z.; Grundke-Iqbal, I. Mechanisms of neurofibrillary degeneration and the formation of neurofibrillary tangles. J. Neural. Transm. Suppl. 1998, 53, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Advances in tau-focused drug discovery for Alzheimer’s disease and related tauopathies. Nat. Rev. Drug Discov. 2009, 8, 783–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.L.; Buist, A.; Soares, A.; Callaerts, K.; Calafate, S.; Stevenaert, F.; Daniels, J.P.; Zoll, B.E.; Crowe, A.; Brunden, K.R.; et al. The Dynamics and Turnover of Tau Aggregates in Cultured Cells: INSIGHTS INTO THERAPIES FOR TAUOPATHIES. J. Biol. Chem. 2016, 291, 13175–13193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatakeyama, S.; Matsumoto, M.; Kamura, T.; Murayama, M.; Chui, D.H.; Planel, E.; Takahashi, R.; Nakayama, K.I.; Takashima, A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J. Neurochem. 2004, 91, 299–307. [Google Scholar] [CrossRef]
- Demishtein, A.; Fraiberg, M.; Berko, D.; Tirosh, B.; Elazar, Z.; Navon, A. SQSTM1/p62-mediated autophagy compensates for loss of proteasome polyubiquitin recruiting capacity. Autophagy 2017, 13, 1697–1708. [Google Scholar] [CrossRef] [Green Version]
- Guthrie, C.R.; Kraemer, B.C. Proteasome inhibition drives HDAC6-dependent recruitment of tau to aggresomes. J. Mol. Neurosci. 2011, 45, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Leyk, J.; Goldbaum, O.; Noack, M.; Richter-Landsberg, C. Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. 2015, 55, 1031–1046. [Google Scholar] [CrossRef]
- Balmik, A.A.; Sonawane, S.K.; Chinnathambi, S. Modulation of Actin network and Tau phosphorylation by HDAC6 ZnF UBP domain. BioRxiv 2019, 702571. [Google Scholar] [CrossRef]
- Bishop, P.; Rocca, D.; Henley, J.M. Ubiquitin C-terminal hydrolase L1 (UCH-L1): Structure, distribution and roles in brain function and dysfunction. Biochem. J. 2016, 473, 2453–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Han, Y.; Yu, Q.; Wang, X.; Wang, S.; Liao, X. UCH-L1 Inhibition Decreases the Microtubule-Binding Function of Tau Protein. J. Alzheimers Dis. 2016, 49, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhang, H.; Li, Y.; Liu, C.; Wang, S.; Liao, X. UCH-L1 Inhibition Suppresses tau Aggresome Formation during Proteasomal Impairment. Mol. Neurobiol. 2017, 55, 3812–3821. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.D.; Ziv, N.E. Recent insights on principles of synaptic protein degradation. F1000Research 2017, 6, 675. [Google Scholar] [CrossRef]
- Marsh, J.; Alifragis, P. Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616–623. [Google Scholar] [CrossRef]
- Zhao, X.L.; Wang, W.A.; Tan, J.X.; Huang, J.K.; Zhang, X.; Zhang, B.Z.; Wang, Y.H.; YangCheng, H.Y.; Zhu, H.L.; Sun, X.J.; et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J. Neurosci. 2010, 30, 1512–1522. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Choi, I.Y.; Michaelis, M.L.; Lee, P. Quantitative in vivo measurement of early axonal transport deficits in a triple transgenic mouse model of Alzheimer’s disease using manganese-enhanced MRI. Neuroimage 2011, 56, 1286–1292. [Google Scholar] [CrossRef] [Green Version]
- Gilley, J.; Seereeram, A.; Ando, K.; Mosely, S.; Andrews, S.; Kerschensteiner, M.; Misgeld, T.; Brion, J.P.; Anderton, B.; Hanger, D.P.; et al. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a "P301L" tau knockin mouse. Neurobiol. Aging 2012, 33, e621. [Google Scholar] [CrossRef]
- Hares, K.; Wilkins, A. Axonal transport proteins as biomarkers of neurodegeneration? Biomark. Med. 2017, 11, 589–591. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.S.P.; Tan, J.M.M.; Soong, W.-E.; Hussein, K.; Nukina, N.; Dawson, V.L.; Dawson, T.M.; Cuervo, A.M.; Lim, K.-L. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum. Mol. Genet. 2008, 17, 2570–2582. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- LaPointe, N.E.; Morfini, G.; Pigino, G.; Gaisina, I.N.; Kozikowski, A.P.; Binder, L.I.; Brady, S.T. The amino terminus of tau inhibits kinesin-dependent axonal transport: Implications for filament toxicity. J. Neurosci. Res. 2009, 87, 440–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pigino, G.; Morfini, G.; Atagi, Y.; Deshpande, A.; Yu, C.; Jungbauer, L.; LaDu, M.; Busciglio, J.; Brady, S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. USA 2009, 106, 5907–5912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolma, K.; Iacobucci, G.J.; Hong Zheng, K.; Shandilya, J.; Toska, E.; White, J.A., 2nd; Spina, E.; Gunawardena, S. Presenilin influences glycogen synthase kinase-3 beta (GSK-3beta) for kinesin-1 and dynein function during axonal transport. Hum. Mol. Genet. 2014, 23, 1121–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaan, N.M.; Pigino, G.F.; Brady, S.T.; Lazarov, O.; Binder, L.I.; Morfini, G.A. Axonal degeneration in Alzheimer’s disease: When signaling abnormalities meet the axonal transport system. Exp. Neurol. 2013, 246, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammineni, P.; Cai, Q. Defective retrograde transport impairs autophagic clearance in Alzheimer disease neurons. Autophagy 2017, 13, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Aboud, O.; Parcon, P.A.; DeWall, K.M.; Liu, L.; Mrak, R.E.; Griffin, W.S. Aging, Alzheimer’s, and APOE genotype influence the expression and neuronal distribution patterns of microtubule motor protein dynactin-P50. Front. Cell Neurosci. 2015, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Funderburk, S.F.; Marcellino, B.K.; Yue, Z. Cell "self-eating" (autophagy) mechanism in Alzheimer’s disease. Mt. Sinai J. Med. 2010, 77, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.Y.; Kuo, P.C.; Wang, Y.T.; Lin, H.T.; Roe, A.D.; Wang, B.Y.; Han, C.L.; Hyman, B.T.; Chen, Y.J.; Tai, H.C. Beta-Amyloid Induces Pathology-Related Patterns of Tau Hyperphosphorylation at Synaptic Terminals. J. Neuropathol. Exp. Neurol. 2018, 77, 814–826. [Google Scholar] [CrossRef]
- Viana da Silva, S.; Haberl, M.G.; Zhang, P.; Bethge, P.; Lemos, C.; Goncalves, N.; Gorlewicz, A.; Malezieux, M.; Goncalves, F.Q.; Grosjean, N.; et al. Early synaptic deficits in the APP/PS1 mouse model of Alzheimer’s disease involve neuronal adenosine A2A receptors. Nat. Commun. 2016, 7, 11915. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Szebenyi, G.; Brown, H.; Pant, H.C.; Pigino, G.; DeBoer, S.; Beffert, U.; Brady, S.T. A novel CDK5-dependent pathway for regulating GSK3 activity and kinesin-driven motility in neurons. EMBO J. 2004, 23, 2235–2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pigino, G.; Morfini, G.; Pelsman, A.; Mattson, M.P.; Brady, S.T.; Busciglio, J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 2003, 23, 4499–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanaan, N.M.; Morfini, G.; Pigino, G.; LaPointe, N.E.; Andreadis, A.; Song, Y.; Leitman, E.; Binder, L.I.; Brady, S.T. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 2012, 33, 826.e15–826.e30. [Google Scholar] [CrossRef] [Green Version]
- Gowrishankar, S.; Wu, Y.; Ferguson, S.M. Impaired JIP3-dependent axonal lysosome transport promotes amyloid plaque pathology. J. Cell Biol. 2017, 216, 3291–3305. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Tammineni, P.; Agrawal, C.; Jeong, Y.Y.; Cai, Q. Autophagy-mediated Regulation of BACE1 Protein Trafficking and Degradation. J. Biol. Chem. 2017, 292, 1679–1690. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Cai, C.Z.; Song, J.X.; Tan, J.Q.; Durairajan, S.S.K.; Iyaswamy, A.; Wu, M.Y.; Chen, L.L.; Yue, Z.; Li, M.; et al. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy 2017, 13, 2028–2040. [Google Scholar] [CrossRef]
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Felzen, V.; Hiebel, C.; Sturner, E.; Perumal, N.; Manicam, C.; Sehn, E.; Grus, F.; Wolfrum, U.; Behl, C. Enhanced autophagic-lysosomal activity and increased BAG3-mediated selective macroautophagy as adaptive response of neuronal cells to chronic oxidative stress. Redox Biol. 2019, 24, 101181. [Google Scholar] [CrossRef]
- Tang, M.; Ji, C.; Pallo, S.; Rahman, I.; Johnson, G.V.W. Nrf2 mediates the expression of BAG3 and autophagy cargo adaptor proteins and tau clearance in an age-dependent manner. Neurobiol. Aging 2018, 63, 128–139. [Google Scholar] [CrossRef]
- Wang, Q.; Li, J.; Yang, X.; Sun, H.; Gao, S.; Zhu, H.; Wu, J.; Jin, W. Nrf2 is associated with the regulation of basal transcription activity of the BRCA1 gene. Acta Biochim. Biophys. Sin. (Shanghai) 2013, 45, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitrov, S.; Brennerova, M.; Forejt, J. Expression profiles and intergenic structure of head-to-head oriented Brca1 and Nbr1 genes. Gene 2001, 262, 89–98. [Google Scholar] [CrossRef]
- Kirkin, V.; Lamark, T.; Sou, Y.S.; Bjorkoy, G.; Nunn, J.L.; Bruun, J.A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanninen, K.; Malm, T.M.; Jyrkkanen, H.K.; Goldsteins, G.; Keksa-Goldsteine, V.; Tanila, H.; Yamamoto, M.; Yla-Herttuala, S.; Levonen, A.L.; Koistinaho, J. Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol. Cell Neurosci. 2008, 39, 302–313. [Google Scholar] [CrossRef]
- Jena, K.K.; Kolapalli, S.P.; Mehto, S.; Nath, P.; Das, B.; Sahoo, P.K.; Ahad, A.; Syed, G.H.; Raghav, S.K.; Senapati, S.; et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef] [Green Version]
- Tanji, K.; Miki, Y.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [Green Version]
- Nicot, A.S.; Lo Verso, F.; Ratti, F.; Pilot-Storck, F.; Streichenberger, N.; Sandri, M.; Schaeffer, L.; Goillot, E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy 2014, 10, 1036–1053. [Google Scholar] [CrossRef] [Green Version]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef]
- Odagiri, S.; Tanji, K.; Mori, F.; Miki, Y.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Brain expression level and activity of HDAC6 protein in neurodegenerative dementia. Biochem. Biophys. Res. Commun. 2013, 430, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schluter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.H.; Xie, L.; Song, S.; Xie, Y.; Allen, L.; Ajit, D.; Hong, J.S.; Chen, X.; Meeker, R.B.; Cohen, T.J. The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep. 2017, 20, 2169–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.J.; Huang, F.I.; Liou, J.P.; Yang, C.R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Sahara, N.; Murayama, M.; Mizoroki, T.; Urushitani, M.; Imai, Y.; Takahashi, R.; Murata, S.; Tanaka, K.; Takashima, A. In vivo evidence of CHIP up-regulation attenuating tau aggregation. J. Neurochem. 2005, 94, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Ferreira, E.; Branca, C.; Oddo, S. p62 improves AD-like pathology by increasing autophagy. Mol. Psychiatry 2017, 22, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Ramesh Babu, J.; Lamar Seibenhener, M.; Peng, J.; Strom, A.L.; Kemppainen, R.; Cox, N.; Zhu, H.; Wooten, M.C.; Diaz-Meco, M.T.; Moscat, J.; et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J. Neurochem. 2008, 106, 107–120. [Google Scholar] [CrossRef]
- Joshi, G.; Gan, K.A.; Johnson, D.A.; Johnson, J.A. Increased Alzheimer’s disease-like pathology in the APP/ PS1DeltaE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol. Aging 2015, 36, 664–679. [Google Scholar] [CrossRef] [Green Version]
- Aviles-Reyes, R.X.; Jonhson, D.; Johnson, J. Astrocytic Overexpression of Nrf2 Leads to Reduced Disease Progression in App/Ps1 Transgenic Mice. Alzheimer’s Dement. 2018, 14, P346–P347. [Google Scholar] [CrossRef]
- Jo, C.; Gundemir, S.; Pritchard, S.; Jin, Y.N.; Rahman, I.; Johnson, G.V. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 2014, 5, 3496. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, R.; Carrera, I.; Xu, S.; Lakshmana, M.K. TFEB Overexpression in the P301S Model of Tauopathy Mitigates Increased PHF1 Levels and Lipofuscin Puncta and Rescues Memory Deficits12. eNeuro 2016, 3. [Google Scholar] [CrossRef]
- Polito, V.A.; Li, H.; Martini-Stoica, H.; Wang, B.; Yang, L.; Xu, Y.; Swartzlander, D.B.; Palmieri, M.; di Ronza, A.; Lee, V.M.Y.; et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014, 6, 1142–1160. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Yan, P.; Ma, X.; Liu, H.; Perez, R.; Zhu, A.; Gonzales, E.; Tripoli, D.L.; Czerniewski, L.; Ballabio, A.; et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Aβ Generation and Amyloid Plaque Pathogenesis. J. Neurosci. 2015, 35, 12137–12151. [Google Scholar] [CrossRef] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [Green Version]
- Sardiello, M. Transcription factor EB: From master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann. N. Y. Acad. Sci. 2016, 1371, 3–14. [Google Scholar] [CrossRef]
- Tsunemi, T.; Ashe, T.D.; Morrison, B.E.; Soriano, K.R.; Au, J.; Roque, R.A.; Lazarowski, E.R.; Damian, V.A.; Masliah, E.; La Spada, A.R. PGC-1alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci. Transl. Med. 2012, 4, 142ra197. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2019, 1–18. [Google Scholar] [CrossRef]
- Sweeney, G.; Song, J. The association between PGC-1alpha and Alzheimer’s disease. Anat. Cell Biol. 2016, 49, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Song, J.X.; Sun, Y.R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.H.; Xu, Z.; Wang, M.Z.; Liu, L.F.; Huang, Y.Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Song, J.-X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.-B.; Tong, B.C.-K.; Iyaswamy, A.; Shang, W.-B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 2019, e13069. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Sato, Y.; Im, E.; Berg, M.; Bordi, M.; Darji, S.; Kumar, A.; Mohan, P.S.; Bandyopadhyay, U.; Diaz, A.; et al. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-betaCTF (C99). J. Neurosci. 2019, 39, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevot, G.; Soria, F.N.; Thiolat, M.L.; Daniel, J.; Verlhac, J.B.; Blanchard-Desce, M.; Bezard, E.; Barthelemy, P.; Crauste-Manciet, S.; Dehay, B. Harnessing Lysosomal pH through PLGA Nanoemulsion as a Treatment of Lysosomal-Related Neurodegenerative Diseases. Bioconjug. Chem. 2018, 29, 4083–4089. [Google Scholar] [CrossRef]
- Baltazar, G.C.; Guha, S.; Lu, W.; Lim, J.; Boesze-Battaglia, K.; Laties, A.M.; Tyagi, P.; Kompella, U.B.; Mitchell, C.H. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS ONE 2012, 7, e49635. [Google Scholar] [CrossRef] [Green Version]
- Mirzoev, T.; Tyganov, S.; Vilchinskaya, N.; Lomonosova, Y.; Shenkman, B. Key Markers of mTORC1-Dependent and mTORC1-Independent Signaling Pathways Regulating Protein Synthesis in Rat Soleus Muscle During Early Stages of Hindlimb Unloading. Cell Physiol. Biochem. 2016, 39, 1011–1020. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rena, G.; Guo, S.; Cichy, S.C.; Unterman, T.G.; Cohen, P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999, 274, 17179–17183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.L.; Zheng, W.; Halloran, J.J.; Burbank, R.R.; Hussong, S.A.; Hart, M.J.; Javors, M.; Shih, Y.Y.; Muir, E.; Solano Fonseca, R.; et al. Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2013, 33, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Fukuda, T.; Christians, U.; Perentesis, J.P.; Fouladi, M.; Vinks, A.A. Population pharmacokinetics of temsirolimus and sirolimus in children with recurrent solid tumours: A report from the Children’s Oncology Group. Br. J. Clin. Pharm. 2017, 83, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Zhang, Q.Q.; Cao, L.; Wang, H.F.; Tan, M.S.; Gao, Q.; Qin, H.; Zhang, Y.D.; et al. Temsirolimus attenuates tauopathy in vitro and in vivo by targeting tau hyperphosphorylation and autophagic clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Zhang, X.; Teng, Z.; Zhang, T.; Li, Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur. J. Pharm. 2014, 740, 312–320. [Google Scholar] [CrossRef]
- Begum, A.N.; Jones, M.R.; Lim, G.P.; Morihara, T.; Kim, P.; Heath, D.D.; Rock, C.L.; Pruitt, M.A.; Yang, F.; Hudspeth, B.; et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J. Pharm. Exp. Ther. 2008, 326, 196–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.; Qiu, S.; Lu, D.X.; Dong, J. Curcumin improves learning and memory ability and its neuroprotective mechanism in mice. Chin. Med. J. 2008, 121, 832–839. [Google Scholar] [CrossRef] [PubMed]
- Beevers, C.S.; Chen, L.; Liu, L.; Luo, Y.; Webster, N.J.; Huang, S. Curcumin disrupts the Mammalian target of rapamycin-raptor complex. Cancer Res. 2009, 69, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.; Kunnumakkara, A.B.; Aggarwal, B.B. Curcumin as “Curecumin”: From kitchen to clinic. Biochem. Pharm. 2008, 75, 787–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvesh, A.S.; Carroll, R.T.; Bishayee, A.; Novotny, N.A.; Geldenhuys, W.J.; Van der Schyf, C.J. Curcumin and neurodegenerative diseases: A perspective. Expert Opin. Investig. Drugs 2012, 21, 1123–1140. [Google Scholar] [CrossRef]
- Rigacci, S.; Miceli, C.; Nediani, C.; Berti, A.; Cascella, R.; Pantano, D.; Nardiello, P.; Luccarini, I.; Casamenti, F.; Stefani, M. Oleuropein aglycone induces autophagy via the AMPK/mTOR signalling pathway: A mechanistic insight. Oncotarget 2015, 6, 35344–35357. [Google Scholar] [CrossRef] [Green Version]
- Grossi, C.; Rigacci, S.; Ambrosini, S.; Ed Dami, T.; Luccarini, I.; Traini, C.; Failli, P.; Berti, A.; Casamenti, F.; Stefani, M. The polyphenol oleuropein aglycone protects TgCRND8 mice against Ass plaque pathology. PLoS ONE 2013, 8, e71702. [Google Scholar] [CrossRef]
- Luccarini, I.; Grossi, C.; Rigacci, S.; Coppi, E.; Pugliese, A.M.; Pantano, D.; la Marca, G.; Ed Dami, T.; Berti, A.; Stefani, M.; et al. Oleuropein aglycone protects against pyroglutamylated-3 amyloid-ss toxicity: Biochemical, epigenetic and functional correlates. Neurobiol. Aging 2015, 36, 648–663. [Google Scholar] [CrossRef]
- Congdon, E.E.; Wu, J.W.; Myeku, N.; Figueroa, Y.H.; Herman, M.; Marinec, P.S.; Gestwicki, J.E.; Dickey, C.A.; Yu, W.H.; Duff, K.E. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy 2012, 8, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeda, Y.; Saito, M.; Maeda, S.; Ishida, K.; Nakamura, A.; Kojima, S.; Takashima, A. Methylene Blue Inhibits Formation of Tau Fibrils but not of Granular Tau Oligomers: A Plausible Key to Understanding Failure of a Clinical Trial for Alzheimer’s Disease. J. Alzheimers Dis. 2019, 68, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Ren, J.; He, X.; Chen, H.; Wei, T.; Feng, W. YWHA/14-3-3 proteins recognize phosphorylated TFEB by a noncanonical mode for controlling TFEB cytoplasmic localization. Autophagy 2019, 15, 1017–1030. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; Facchinetti, V.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef]
- Schaeffer, V.; Lavenir, I.; Ozcelik, S.; Tolnay, M.; Winkler, D.T.; Goedert, M. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain 2012, 135, 2169–2177. [Google Scholar] [CrossRef] [Green Version]
- Kruger, U.; Wang, Y.; Kumar, S.; Mandelkow, E.M. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol. Aging 2012, 33, 2291–2305. [Google Scholar] [CrossRef]
- Tien, N.T.; Karaca, I.; Tamboli, I.Y.; Walter, J. Trehalose Alters Subcellular Trafficking and the Metabolism of the Alzheimer-associated Amyloid Precursor Protein. J. Biol. Chem. 2016, 291, 10528–10540. [Google Scholar] [CrossRef] [Green Version]
- Portbury, S.D.; Hare, D.J.; Sgambelloni, C.; Perronnes, K.; Portbury, A.J.; Finkelstein, D.I.; Adlard, P.A. Trehalose Improves Cognition in the Transgenic Tg2576 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 60, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Liang, Y.; Xu, F.; Sun, B.; Wang, Z. Trehalose rescues Alzheimer’s disease phenotypes in APP/PS1 transgenic mice. J. Pharm. Pharm. 2013, 65, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharm. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Song, H.L.; Demirev, A.V.; Kim, N.Y.; Kim, D.H.; Yoon, S.Y. Ouabain activates transcription factor EB and exerts neuroprotection in models of Alzheimer’s disease. Mol. Cell Neurosci. 2019, 95, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, M.; Ding, X.; Yan, C.; Song, Z.; Chen, L.; Huang, X.; Wang, X.; Jian, Y.; Tang, G.; et al. Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 2016, 18, 1065–1077. [Google Scholar] [CrossRef]
- Chandra, S.; Roy, A.; Jana, M.; Pahan, K. Cinnamic acid activates PPARalpha to stimulate Lysosomal biogenesis and lower Amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol. Dis. 2019, 124, 379–395. [Google Scholar] [CrossRef]
- Chandra, S.; Jana, M.; Pahan, K. Aspirin Induces Lysosomal Biogenesis and Attenuates Amyloid Plaque Pathology in a Mouse Model of Alzheimer’s Disease via PPARalpha. J. Neurosci. 2018, 38, 6682–6699. [Google Scholar] [CrossRef]
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS ONE 2013, 8, e62459. [Google Scholar] [CrossRef] [Green Version]
- Frederick, C.; Ando, K.; Leroy, K.; Heraud, C.; Suain, V.; Buee, L.; Brion, J.P. Rapamycin ester analog CCI-779/Temsirolimus alleviates tau pathology and improves motor deficit in mutant tau transgenic mice. J. Alzheimers Dis. 2015, 44, 1145–1156. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Liu, M.Y.; Wang, F.; Wei, M.J.; Wang, S.; Wu, C.F.; Yang, J.Y. Anti-amnesic effect of pseudoginsenoside-F11 in two mouse models of Alzheimer’s disease. Pharm. Biochem. Behav. 2013, 106, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, J.; Liu, C.; Xie, J.; Qiu, S.; Yang, X.; Wu, C. Pseudoginsenoside-F11 alleviates cognitive deficits and Alzheimer’s disease-type pathologies in SAMP8 mice. Pharm. Res. 2019, 139, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2019, 25, 148–167. [Google Scholar] [CrossRef] [Green Version]
- Neff, E.P. Animal models of Alzheimer’s disease embrace diversity. Lab. Anim. 2019, 48, 255–259. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound | Mode of Action | Animal Model | Dose/Route | Result | Reference |
|---|---|---|---|---|---|
| Curcumin | mTOR inhibition |
|
|
| [130,150] |
| Rapamycin | mTOR inhibition |
|
|
| [144,146,180] |
| Temsirolimus | mTOR inhibition |
|
|
| [148,181] |
| Oleuropein | AMPK activation, mTOR inhibition |
|
|
| [159] |
| Methylene blue | mTOR inhibition |
|
|
| [161] |
| Ouabain | mTOR inhibition, TFEB activation |
|
|
| [176] |
| Trehalose | TFEB activation |
|
|
| [170,173,174] |
| Pseudoginsenoside-F11 (PF-11) | TFEB activation |
|
|
| [182,183] |
| Hep-14 | TFEB activation |
|
|
| [177] |
| Aspirin | PPARGC1α mediated TFEB activation |
|
|
| [179] |
| Cinnamic acid | PPARGC1α mediated TFEB activation |
|
|
| [178] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malampati, S.; Song, J.-X.; Chun-Kit Tong, B.; Nalluri, A.; Yang, C.-B.; Wang, Z.; Gopalkrishnashetty Sreenivasmurthy, S.; Zhu, Z.; Liu, J.; Su, C.; et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells 2020, 9, 311. https://doi.org/10.3390/cells9020311
Malampati S, Song J-X, Chun-Kit Tong B, Nalluri A, Yang C-B, Wang Z, Gopalkrishnashetty Sreenivasmurthy S, Zhu Z, Liu J, Su C, et al. Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells. 2020; 9(2):311. https://doi.org/10.3390/cells9020311
Chicago/Turabian StyleMalampati, Sandeep, Ju-Xian Song, Benjamin Chun-Kit Tong, Anusha Nalluri, Chuan-Bin Yang, Ziying Wang, Sravan Gopalkrishnashetty Sreenivasmurthy, Zhou Zhu, Jia Liu, Chengfu Su, and et al. 2020. "Targeting Aggrephagy for the Treatment of Alzheimer’s Disease" Cells 9, no. 2: 311. https://doi.org/10.3390/cells9020311
APA StyleMalampati, S., Song, J. -X., Chun-Kit Tong, B., Nalluri, A., Yang, C. -B., Wang, Z., Gopalkrishnashetty Sreenivasmurthy, S., Zhu, Z., Liu, J., Su, C., Krishnamoorthi, S., Iyaswamy, A., Cheung, K. -H., Lu, J. -H., & Li, M. (2020). Targeting Aggrephagy for the Treatment of Alzheimer’s Disease. Cells, 9(2), 311. https://doi.org/10.3390/cells9020311