miRNAs as Key Players in the Management of Cutaneous Melanoma


Abstract
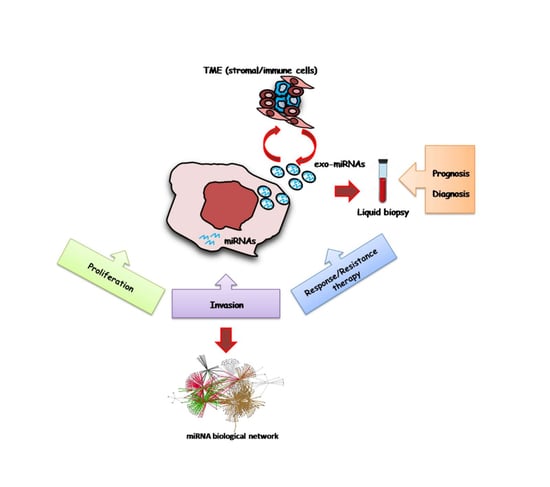
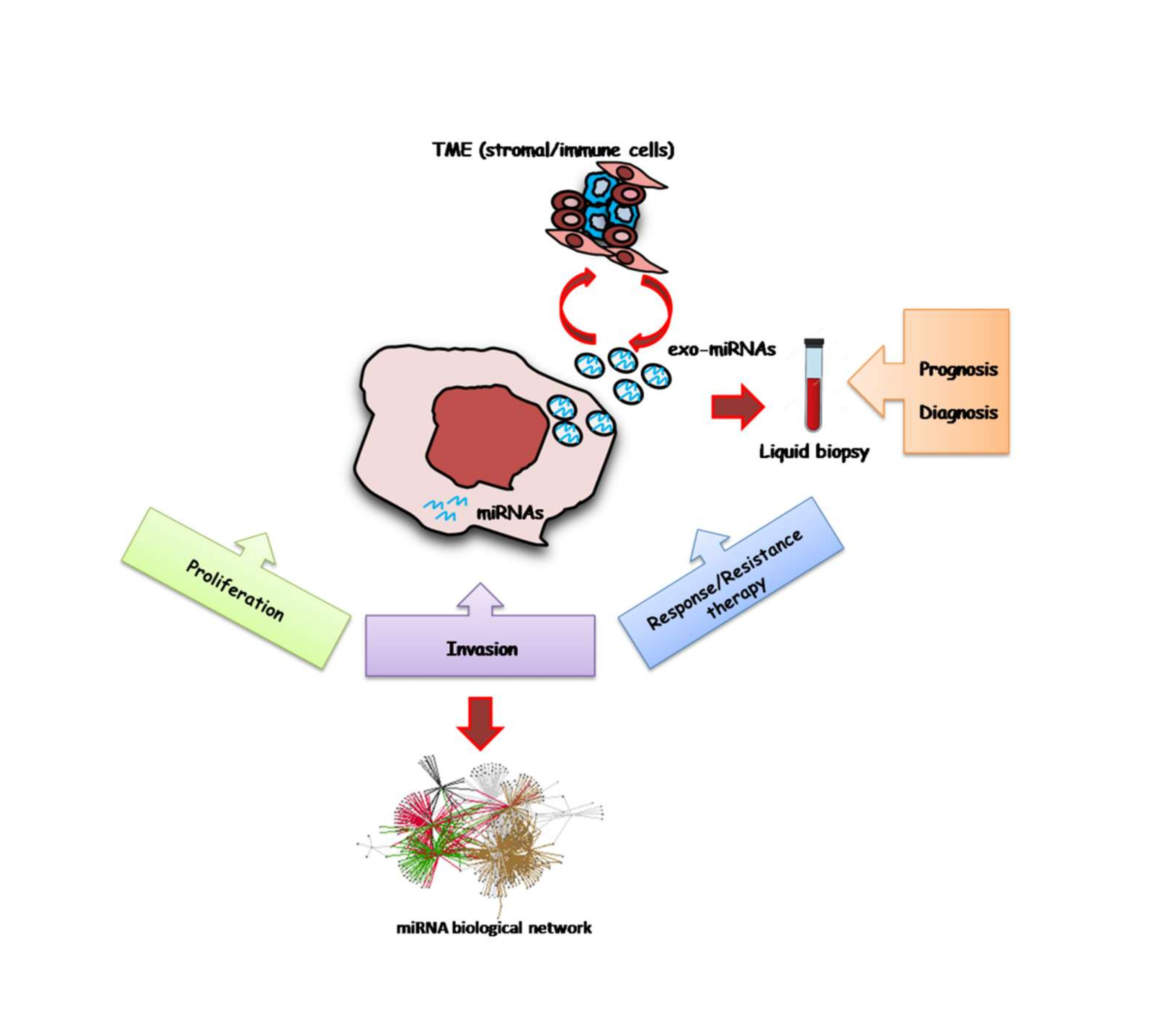
:
1. Introduction
2. State of the Art of Targeted Therapy Options
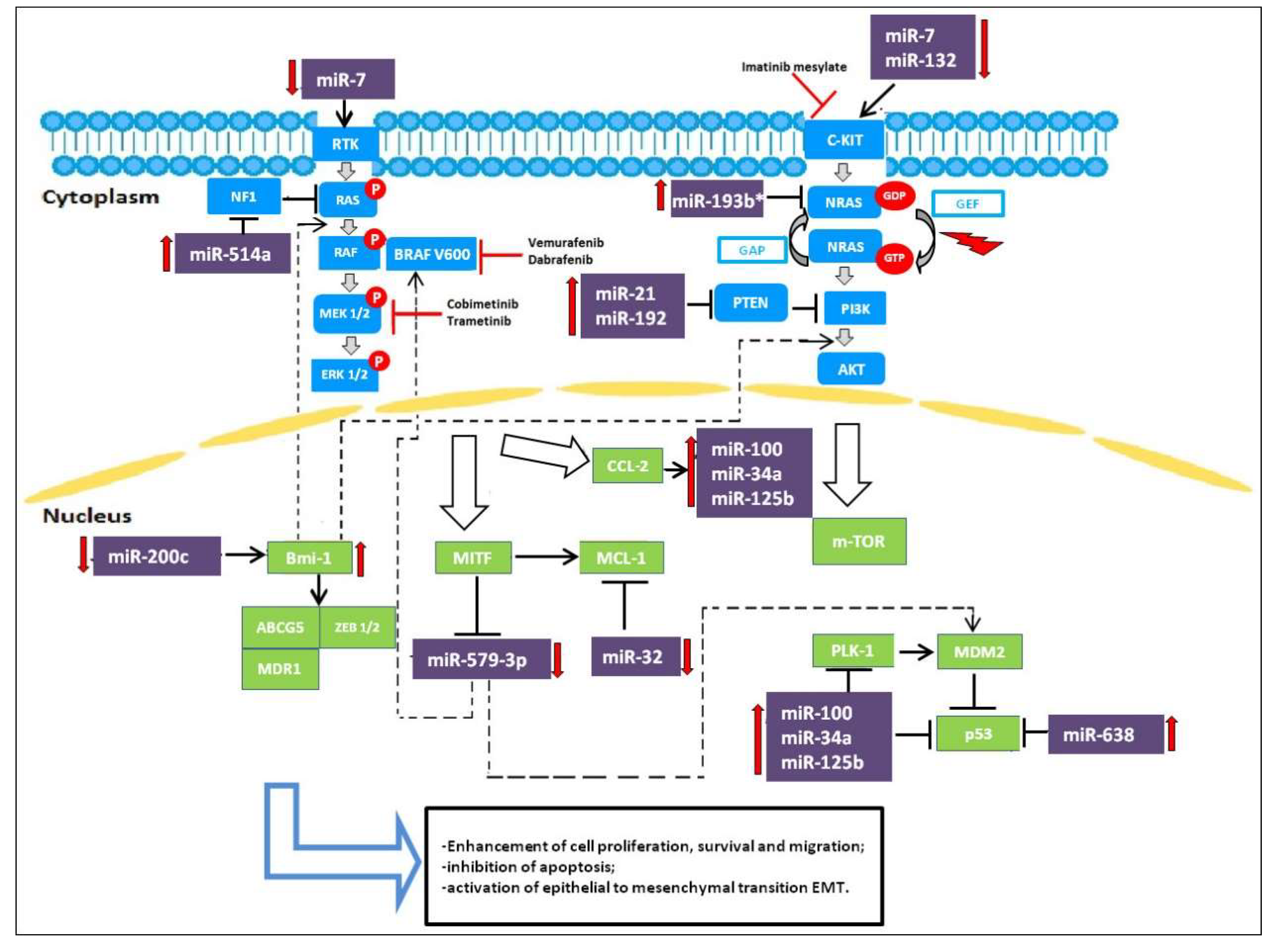
2.1. miRNAs Acting as Oncosuppressors in Melanoma
2.2. miRNAs Acting as Oncogenes
2.3. miRNA-Based Therapeutic Approaches
- (a)
- miRNA inhibition therapy with anti-miRNA oligomers, which are 17–22 nucleotides long, single-stranded, chemically modified, competitive inhibitors of miRNAs, causing upregulation of the target mRNA;
- (b)
- Synthetic miRNA mimetic agents used as replacement therapy to replace or substitute lost miRNA;
- (c)
- Small-molecule inhibitors of miRNA (SMIRs) to either inhibit miRNA biogenesis or impede the interaction of miRNA with its target;
- (d)
- miRNAs targeted during transport within the tumor milieu or to distant sites in microvesicles and exosomes [34].
3. The Advent of Immune Checkpoint Inhibitors: Do miRNAs Have a Role?
4. miRNAs and the Interplay with Tumor Microenvironment
5. Circulating MicroRNA Biomarkers in Melanoma
Exo-miRNAs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matthews, N.H.; Li, W.Q.; Qureshi, A.A.; Weinstock, M.A.; Cho, E. Epidemiology of Melanoma. In Cutaneous Melanoma: Etiology and Therapy; Ward, W.H., Farma, J.M., Eds.; Codon Publications: Brisbane, Austrilia, 2017. [Google Scholar]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 2015, 386, 444–451. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Buder-Bakhaya, K.; Machiraju, D.; Hassel, J.C. Liquid biopsy: Value for melanoma therapy. Oncol. Res. Treat. 2017, 40, 430–434. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Flaherty, K.T. Resistance to BRAF-targeted therapy in melanoma. Eur. J. Cancer 2013, 49, 1297–1304. [Google Scholar] [CrossRef]
- Bello, D.M.; Ariyan, C.E.; Carvajal, R.D. Melanoma mutagenesis and aberrant cell signaling. Cancer Control 2013, 20, 261–281. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. Immuno Targets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, K.T. Targeting Metastatic Melanoma. Annu. Rev. Med. 2012, 63, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Riveiro-Falkenbach, E.; Villanueva, C.A.; Garrido, M.C.; Ruano, Y.; Garcìa-Martìn, R.M.; Godoy, E.; Ortiz-Romero, P.L.; Rìos-Martìn, J.J.; Santos-Briz, A.; Rodrìguez-Peralto, J.L. Intra- and inter-tumoral homogeneity BRAFV600E mutations in melanoma tumors. J. Investig. Dermatol. 2015, 135, 3078–3085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; García, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco. Targets. Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.L.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef]
- Babaei, M.A.; Kamalidehghan, B.; Saleem, M.; Huri, H.Z.; Ahmadipour, F. Receptor tyrosine kinase (c-Kit) inhibitors: A potential therapeutic target in cancer cells. Drug Des. Devel. Ther. 2016, 10, 2443–2459. [Google Scholar] [CrossRef] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Mishra, P.J.; Merlino, G. Integrated Genomics Identifies miR-32/MCL-1 Pathway as a Critical Driver of Melanomagenesis: Implications for miR-Replacement and Combination Therapy. PLoS ONE 2016, 11, e0165102. [Google Scholar] [CrossRef]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Caponea, M.; et al. miR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Cui, R.; Xu, X. MiR-200c inhibits melanoma progression and drug resistance through down-regulation of Bmi-1. Am. J. Pathol. 2012, 181, 1823–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Wang, T.; Yang, R.; Xie, L.; Zhang, G.; Krepler, C.; Xiao, M.; Beqiri, M.; Xu, W.; et al. miR-200c/Bmi1 axis and epithelial-mesenchymal transition contribute to acquired resistance to BRAF inhibitor treatment. Pigment Cell Melanoma Res. 2015, 28, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, J.; Sun, Y.; Zhang, Y.; Dong, L.; Shen, C.; Yang, L.; Yang, M.; Li, Y.; Shen, G.; et al. miR-7 reverses the resistance to BRAFi in melanoma by targeting EGFR/IGF-1R/CRAF and inhibiting the MAPK and PI3K/AKT signaling pathways. Oncotarget 2016, 7, 53558–53570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Chen, M.; Wang, Y.; Cui, P.; Liu, S.; Xu, Z. MicroRNA-21 regulates the ERK/NF-kB signalingpathway to affect the proliferation, migration, and apoptosis of human melanoma A375 cells by targeting SPRY1, PDCD4, and PTEN. Mol. Carcinog. 2017, 56, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Lv, X.; Li, J.; Li, J.; Li, X.; Li, W.; Li, Y. The status of microRNA-21 expression and its clinical significance in human cutaneous malignant melanoma. Acta Histochem. 2012, 114, 582–588. [Google Scholar] [CrossRef]
- Fattore, L.; Costantini, S.; Malpicci, D.; Ruggiero, C.F.; Ascierto, P.A.; Croce, C.M.; Mancini, R.; Ciliberto, G. MicroRNAs in melanoma development and resistance to target therapy. Oncotarget 2017, 8, 22262–22278. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, A.; Schmitz, U.; Raatz, Y.; Schönherr, M.; Kottek, T.; Schauer, M.; Franz, S.; Saalbach, A.; Anderegg, U.; Wolkenhauer, O.; et al. miR-638 promotes melanoma metastasis and protects melanoma cells from apoptosis and autophagy. Oncotarget 2015, 6, 2966–2980. [Google Scholar] [CrossRef] [Green Version]
- Vergani, E.; Di Guardo, L.; Dugo, M.; Rigoletto, S.; Tragni, G.; Ruggeri, R.; Perrone, F.; Tamborini, E.; Gloghini, A.; Arienti, F.; et al. Overcoming melanoma resistance to vemurafenib by targeting CCL2-induced miR-34a, miR-100 and miR-125b. Oncotarget 2016, 7, 4428–4441. [Google Scholar] [CrossRef] [Green Version]
- Pinto, R.; Strippoli, S.; De Summa, S.; Albano, A.; Azzariti, A.; Guida, G.; Popescu, O.; Lorusso, V.; Guida, M.; Tommasi, S. MicroRNA expression in BRAF-mutated and wild-type metastatic melanoma and its correlation with response duration to BRAF inhibitors. Expert Opin. Ther. Targets 2015, 19, 1027–1035. [Google Scholar] [CrossRef]
- Stark, M.S.; Bonazzi, V.F.; Boyle, G.M.; Palmer, J.M.; Symmons, J.; Lanagan, C.M.; Schmidt, C.W.; Herington, A.C.; Ballotti, R.; Pollock, P.M.; et al. miR-514a regulates the tumour suppressor NF1 and modulates BRAFi sensitivity in melanoma. Oncotarget 2015, 6, 17753–17763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Stenvang, J.; Petri, A.; Lindow, M.; Obad, S.; Kauppinen, S. Inhibition of microRNA function by antimiR oligonucleotides. Silence 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Long, G.V.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Long-term outcomes in patients (pts) with ipilimumab (ipi)-naive advanced melanoma in the phase 3 KEYNOTE-006 study who completed pembrolizumab (pembro) treatment. J. Clin. Oncol. 2017, 35, 9504. [Google Scholar] [CrossRef]
- Balch, C.M.; Gershenwald, J.E.; Soong, S.J.; Thompson, J.F.; Atkins, M.B.; Byrd, D.R.; Buzaid, A.C.; Cochran, A.J.; Coit, D.G.; Ding, S.; et al. Final version of 2009 AJCC melanoma staging and classification. J. Clin. Oncol. 2009, 27, 6199–6206. [Google Scholar] [CrossRef] [Green Version]
- Kelderman, S.; Heemskerk, B.; Van Tinteren, H.; Van Den Brom, R.R.H.; Hospers, G.A.P.; Van Den Eertwegh, A.J.M.; Kapiteijn, E.W.; De Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 63, 449–458. [Google Scholar] [CrossRef]
- Nosrati, A.; Tsai, K.K.; Goldinger, S.M.; Tumeh, P.; Grimes, B.; Loo, K.; Algazi, A.P.; Nguyen-Kim, T.D.L.; Levesque, M.; Dummer, R.; et al. Evaluation of clinicopathological factors in PD-1 response: Derivation and validation of a prediction scale for response to PD-1 monotherapy. Br. J. Cancer 2017, 116, 1141–1147. [Google Scholar] [CrossRef] [Green Version]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for response of melanoma patients to immune checkpoint inhibitors: A systematic review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef]
- Diem, S.; Kasenda, B.; Spain, L.; Martin-Liberal, J.; Marconcini, R.; Gore, M.; Larkin, J. Serum lactate dehydrogenase as an early marker for outcome in patients treated with anti-PD-1 therapy in metastatic melanoma. Br. J. Cancer 2016, 114, 256–261. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015, 350, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, 3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Kendall, S.M.; Makarov, V.; Walsh, L.A.; Desrichard, A.; Weinhold, N.; Chan, T.A. Recurrent SERPINB3 and SERPINB4 mutations in patients who respond to anti-CTLA4 immunotherapy. Nat. Genet. 2016, 48, 1327–1329. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.; Chen, B.; Fu, X.; Calin, G.A. Key questions about the checkpoint blockade-are microRNAs an answer? Cancer Biol. Med. 2018, 15, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Wan, C.; Dong, Z.B.; Wang, B.H.; Liu, H.Y.; Li, Y. Integrative Analysis of Long Noncoding RNA (lncRNA), microRNA (miRNA) and mRNA Expression and Construction of a Competing Endogenous RNA (ceRNA) Network in Metastatic Melanoma. Med. Sci. Monit. 2019, 25, 2896–2907. [Google Scholar] [CrossRef]
- Xu, H.; Cheung, I.Y.; Guo, H.F.; Cheung, N.K.V. MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: Potential implications for immune based therapy of human solid tumors. Cancer Res. 2009, 69, 6275–6281. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chong, K.K.; Nakamura, Y.; Nguyen, L.; Huang, S.K.; Kuo, C.; Zhang, W.; Yu, H.; Morton, D.L.; Hoon, D.S.B. B7-H3 associated with tumor progression and epigenetic regulatory activity in cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 2050–2058. [Google Scholar] [CrossRef] [Green Version]
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 Is a Negative Regulator of T Cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Suh, W.K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A.; et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Kushner, B.H.; Modak, S.; Pandit-Taskar, N.; Smith-Jones, P.; Zanzonico, P.; Humm, J.L.; Xu, H.; Wolden, S.L.; Souweidane, M.M.; et al. Compartmental intrathecal radioimmunotherapy: Results for treatment for metastatic CNS neuroblastoma. J. Neurooncol. 2010, 97, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekle, C.; Nygren, M.K.; Chen, Y.W.; Dybsjord, I.; Nesland, J.M.; Mælandsmo, G.M.; Fodstad, Ø. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Gabriel, S.S.; Liao, Y.; Gloury, R.; Preston, S.; Henstridge, D.C.; Pellegrini, M.; Zehn, D.; Berberich-Siebelt, F.; Febbraio, M.A.; et al. Transcription Factor IRF4 Promotes CD8+ T Cell Exhaustion and Limits the Development of Memory-like T Cells during Chronic Infection. Immunity 2017, 47, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Usatorre, A.; Sempere, L.F.; Carmona, S.J.; Carretero-Iglesia, L.; Monnot, G.; Speiser, D.E.; Rufer, N.; Donda, A.; Zehn, D.; Jandus, C.; et al. MicroRNA-155 expression is enhanced by T-cell receptor stimulation strength and correlates with improved tumor control in Melanoma. Cancer Immunol. Res. 2019, 7, 1013–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Pagliano, O.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Olive, D.; Kuchroo, V.; Zarour, H.M. CD8 + T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012, 72, 887–896. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audrito, V.; Serra, S.; Stingi, A.; Orso, F.; Gaudino, F.; Bologna, C.; Neri, F.; Garaffo, G.; Nassini, R.; Baroni, G.; et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 2017, 8, 15894–15911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaun, B.; Yamamoto, T.; Badran, B.; Tsunetsugu-Yokota, Y.; Roux, A.; Baitsch, L.; Rouas, R.; Fayyad-Kazan, H.; Baumgaertner, P.; Devevre, E.; et al. Differentiation associated regulation of microRNA expression in vivo in human CD8+T cell subsets. J. Transl. Med. 2011, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Zhang, P.; Krishna, S.; Wang, J.; Lin, X.; Huang, H.; Xie, D.; Gorentla, B.; Huang, R.; Gao, J.; et al. Unexpected positive control of NFκB and miR-155 by DGKa and ζ ensures effector and memory CD8+ T Cell differentiation. Oncotarget 2016, 7, 33744–33764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Q.; Wang, X.; McBride, J.; Fewell, C.; Flemington, E. B-cell receptor activation induces BIC/miR-155 expression through a conserved AP-1 element. J. Biol. Chem. 2008, 283, 2654–2662. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Mazar, J.; Qi, F.; Lee, B.; Marchica, J.; Govindarajan, S.; Shelley, J.; Li, J.-L.; Ray, A.; Perera, R.J. MicroRNA 211 Functions as a Metabolic Switch in Human Melanoma Cells. Mol. Cell. Biol. 2016, 36, 1090–1108. [Google Scholar] [CrossRef] [Green Version]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Pencheva, N.; Tran, H.; Buss, C.; Huh, D.; Drobnjak, M.; Busam, K.; Tavazoie, S.F. Convergent multi-miRNA targeting of ApoEgrives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell 2012, 151, 1068–1082. [Google Scholar] [CrossRef] [Green Version]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the angiogenic switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Noman, M.Z.; Buart, S.; Romero, P.; Ketari, S.; Janji, B.; Mari, B.; Mami-Chouaib, F.; Chouaib, S. Hypoxia-inducible miR-210 regulates the susceptibility of tumor cells to lysis by cytotoxic T cells. Cancer Res. 2012, 72, 4629–4641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathsyaraja, H.; Thies, K.; Taffany, D.A.; Deighan, C.; Liu, T.; Yu, L.; Fernandez, S.A.; Shapiro, C.; Otero, J.; Timmers, C.; et al. CSF1-ETS2-induced microRNA in myeloid cells promote metastatic tumor growth. Oncogene 2015, 34, 3651–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinemann, A.; Zhao, F.; Pechlivanis, S.; Eberle, J.; Steinle, A.; Diederichs, S.; Schadendorf, D.; Paschen, A. Tumor suppressive microRNAs miR-34a/c control cancer cell expression of ULBP2, a stress-induced ligand of the natural killer cell receptor NKG2D. Cancer Res. 2012, 72, 460–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelec, G.; Verschoor, C.P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Not only in tumor immunity. Front. Immunol. 2019, 10, 1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, L.; Fan, J.; Ye, C.; Dominguez, D.; Zhang, Y.; Curiel, T.J.; Fang, D.; Kuzel, T.M.; Zhang, B. Host miR155 promotes tumor growth through a myeloid-derived suppressor cell-dependent mechanism. Cancer Res. 2015, 75, 519–531. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 Is Required for the Accumulation and Functions of Tumor-Expanded Myeloid-Derived Suppressor Cells via Targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; sborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [Green Version]
- Skourti, E.; Logotheti, S.; Kontos, C.K.; Pavlopoulou, A.; Dimoragka, P.T.; Trougakos, I.P.; Gorgoulis, V.; Scorilas, A.; Michalopoulos, I.; Zoumpourlis, V. Progression of mouse skin carcinogenesis is associated with the orchestrated deregulation of mir-200 family members, mir-205 and their common targets. Mol. Carcinog. 2016, 55, 1229–1242. [Google Scholar] [CrossRef]
- Liu, S.; Kumar, S.M.; Lu, H.; Liu, A.; Yang, R.; Pushparajan, A.; Guo, W.; Xu, X. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-κB1-Snail1 pathway in melanoma. J. Pathol. 2012, 226, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2014, 10, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asangani, I.A.; Harms, P.W.; Dodson, L.; Pandhi, M.; Kunju, L.P.; Maher, C.A.; Fullen, D.R.; Johnson, T.M.; Giordano, T.J.; Palanisamy, N.; et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012, 3, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zingg, D.; Debbache, J.; Schaefer, S.M.; Tuncer, E.; Frommel, S.C.; Cheng, P.; Arenas-Ramirez, N.; Haeusel, J.; Zhang, Y.; Bonalli, M.; et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat. Commun. 2015, 6, 6051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Sole, C.; Arnaiz, E.; Manterola, L.; Otaegui, D.; Lawrie, C.H. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin. Cancer Biol. 2019, 58, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular organelles important in intercellular communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Mumford, S.L.; Towler, B.P.; Pashler, A.L.; Gilleard, O.; Martin, Y.; Newbury, S.F. Circulating microRNA biomarkers in melanoma: Tools and challenges in personalised medicine. Biomolecules 2018, 8, E21. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.Y.; Lee, J.H.; Diefenbach, R.J.; Kefford, R.F.; Rizos, H. Liquid biomarkers in melanoma: Detection and discovery. Mol. Cancer 2018, 17, 8. [Google Scholar] [CrossRef]
- Sohel, M.H. Extracellular/Circulating MicroRNAs: Release Mechanisms, Functions and Challenges. Achiev. Life Sci. 2016, 10, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Carpi, S.; Polini, B.; Fogli, S.; Nieri, P.; Romanini, A. Circulating MicroRNAs in Cutaneous Melanoma Diagnosis and Prognosis. Manag. Malig. Melanoma. 2016, 12. [Google Scholar]
- Leidinger, P.; Keller, A.; Borries, A.; Reichrath, J.; Rass, K.; Jager, S.U.; Lenhof, H.P.; Meese, E. High-throughput miRNA profiling of human melanoma blood samples. BMC Cancer 2010, 10, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogli, S.; Polini, B.; Carpi, S.; Pardini, B.; Naccarati, A.; Dubbini, N.; Lanza, M.; Breschi, M.C.; Romanini, A.; Nieri, P. Identification of plasma microRNAs as new potential biomarkers with high diagnostic power in human cutaneous melanoma. Tumor Biol. 2017, 39, 1010428317701646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; He, Q.Y.; Luo, C.Q.; Qian, L.Y. Circulating miR-221 expression level and prognosis of cutaneous malignant melanoma. Med. Sci. Monit. 2014, 20, 2472–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The Prognostic and Predictive Value of Melanoma-related MicroRNAs Using Tissue and Serum: A MicroRNA Expression Analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Polini, B.; Carpi, S.; Romanini, A.; Breschi, M.C.; Nieri, P.; Podestà, A. Circulating cell-free microRNAs in cutaneous melanoma staging and recurrence or survival prognosis. Pigment Cell Melanoma Res. 2019, 32, 486–499. [Google Scholar] [CrossRef]
- Kanemaru, H.; Fukushima, S.; Yamashita, J.; Honda, N.; Oyama, R.; Kakimoto, A.; Masuguchi, S.; Ishihara, T.; Inoue, Y.; Jinnin, M.; et al. The circulating microRNA-221 level in patients with malignant melanoma as a new tumor marker. J. Dermatol. Sci. 2011, 61, 187–193. [Google Scholar] [CrossRef]
- Fleming, N.H.; Zhong, J.; Da Silva, I.P.; De Miera, E.V.S.; Brady, B.; Han, S.W.; Hanniford, D.; Wang, J.; Shapiro, R.L.; Hernando, E.; et al. Serum-based miRNAs in the prediction and detection of recurrence in melanoma patients. Cancer 2015, 121, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Friedman, E.B.; Shang, S.; de Miera, E.V.; Fog, J.U.; Teilum, M.W.; Ma, M.W.; Berman, R.S.; Shapiro, R.L.; Pavlick, A.C.; Hernando, E.; et al. Serum microRNAs as biomarkers for recurrence in melanoma. J. Transl. Med. 2012, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Liu, T.; Qiao, L.; Gao, M.; Li, J. Decreased serum microRNA-206 level predicts unfavorable prognosis in patients with melanoma. Int. J. Clin. Exp. Pathol. 2015, 8, 3097–3103. [Google Scholar] [CrossRef]
- Margue, C.; Reinsbach, S.; Philippidou, D.; Beaume, N.; Walters, C.; Schneider, J.G.; Nashan, D.; Behrmann, I.; Kreis, S. Comparison of a healthy miRNome with melanoma patient miRNomes: Are microRNAs suitable serum biomarkers for cancer? Oncotarget 2015, 6, 12110–12127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiiyama, R.; Fukushima, S.; Jinnin, M.; Yamashita, J.; Miyashita, A.; Nakahara, S.; Kogi, A.; Aoi, J.; Masuguchi, S.; Inoue, Y.; et al. Sensitive detection of melanoma metastasis using circulating microRNA expression profiles. Melanoma Res. 2013, 23, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, R.; Lincoln, M.; Van Laar, B. Development and validation of a plasmabased melanoma biomarker suitable for clinical use. Br. J. Cancer 2018, 118, 857–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karin-Kujundzic, V.; Sola, I.M.; Predavec, N.; Potkonjak, A.; Somen, E.; Mioc, P.; Serman, A.; Vranic, S.; Serman, L. Novel Epigenetic Biomarkers in Pregnancy-Related Disorders and Cancers. Cells 2019, 8, 1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Shen, Q.; Yang, X.; Qiu, Y.; Zhang, W. The role of extracellular vesicles: An epigenetic view of the cancer microenvironment. Biomed Res. Int. 2015, 1, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and biological characterization of exosomes containing prominin-1/CD133. Mol. Cancer 2013, 12, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre, E.; Sanmamed, M.F.; Rodriguez, C.; Carranza, O.; Martín-Algarra, S.; González, Á. Study of circulating MicroRNA-125b levels in serum exosomes in advanced melanoma. Arch. Pathol. Lab. Med. 2014, 138, 828–832. [Google Scholar] [CrossRef]
- Tengda, L.; Shuping, L.; Mingli, G.; Jie, G.; Yun, L.; Weiwei, Z.; Anmei, D. Serum exosomal microRNAs as potent circulating biomarkers for melanoma. Melanoma Res. 2018, 28, 295–303. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Main Deregulated miRNA | Body Fluid Type | Role | Detection Method | Reference |
|---|---|---|---|---|
| miR-186, let-7d*, miR-18a*, miR-145, miR-99a, miR-664, miR-501-5p, miR-378*, miR-29c*, miR-1280, miR-365, miR-1249, miR-328, miR-422a, miR-30d, miR-17* | Blood | Prognostic | microarray/qRT-PCR | [93] |
| miR-15b-5p, miR-149-3p, miR-150-5p, miR-193a-3p, miR-524-5p | Plasma | Prognostic | qRT-PCR | [94] |
| miR-221-3p | Serum | Prognostic | qRT-PCR | [95,98] |
| MELmiR-17 panel (hsa-miR-211, miR-508-3p, miR-514a, miR-4731, miR-146a, miR-509-3p, miR-506-3p, miR-509-5p, miR-508-5p, miR-4487, miR-16, miR-204, miR-513c, miR-513b, miR-145, miR-363-3p, miR-4706) and MELmiR-7 panel (miR-16, miR-211-5p, miR-4487, miR-4706, miR-4731, miR-509-3p, miR-509-5p) | FFPE tissue and serum | Prognostic and Predictive | qRT-PCR | [96] |
| miR-150, miR-15b, miR-425, miR-30d | Serum | Prognostic and Predictive | qRT-PCR | [99] |
| miR-150, miR -15b, miR -199a-5p, miR-33a, miR-424 | Serum | Prognostic and Predictive | qRT-PCR | [100] |
| miR-206 | Serum | Prognostic and Predictive | qRT-PCR | [101] |
| miRNome including miR-193b-3p and miR-720 | Serum, whole blood samples, melanoma tissue, primary melanocyte and keratinocyte cell lines | Prognostic and Predictive | qPCR arrays | [102] |
| miR-9, miR-145, miR-150, miR-155, miR-203, and miR-205 | Serum | Prognostic | qRT-PCR | [103] |
| 38-miRNA signature (MEL38) and 18-miRNA signature (MEL18) | Plasma | Prognostic and Predictive | microarray | [104] |
| miR-216b, miR-889, miR-4307, miR-4272, miR-203, miR-4289, miR-3149, miR-203, miR-3145, miR-1911, miR-513a-3p, miR-3916, miR-886-3p, miR-1182, miR-3613-5p, let-7i, miR-3132, miR-3914, miR-3618, miR-1307, miR-3614-3p, miR-3160, miR-519c-3p, miR-3153, miR-4278, miR-3646, miR-3926, miR-515-5p, miR-3169, miR-10a, miR-140-5p, miR-3148, miR-4271, miR-627, miR-548d-3p, miR-3613-3p, miR-481, miR-571, miR-4274, miR-4277, miR-3686, miR-3074, miR-95, miR-590-3p, miR-525-5p, miR-548g, miR-365, miR-525-3p, miR-320d | exosomes | Prognostic | microarray | [107] |
| miR-16, miR-125b | serum exosomes | Prognostic | qRT-PCR | [108] |
| miR-532-5p, miR-106b, miR-200c, miR-199a-5p, miR-210 | serum exosomes | Prognostic and Predictive | qRT-PCR | [109] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorusso, C.; De Summa, S.; Pinto, R.; Danza, K.; Tommasi, S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells 2020, 9, 415. https://doi.org/10.3390/cells9020415
Lorusso C, De Summa S, Pinto R, Danza K, Tommasi S. miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells. 2020; 9(2):415. https://doi.org/10.3390/cells9020415
Chicago/Turabian StyleLorusso, Celeste, Simona De Summa, Rosamaria Pinto, Katia Danza, and Stefania Tommasi. 2020. "miRNAs as Key Players in the Management of Cutaneous Melanoma" Cells 9, no. 2: 415. https://doi.org/10.3390/cells9020415
APA StyleLorusso, C., De Summa, S., Pinto, R., Danza, K., & Tommasi, S. (2020). miRNAs as Key Players in the Management of Cutaneous Melanoma. Cells, 9(2), 415. https://doi.org/10.3390/cells9020415