c-Jun, Foxo3a, and c-Myc Transcription Factors are Key Regulators of ATP-Mediated Angiogenic Responses in Pulmonary Artery Vasa Vasorum Endothelial Cells † †


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cultures of VVEC
2.2. siRNA Transfection
2.3. Preparation of Nuclear Fraction
2.4. Analysis of Transcription Factor Transactivation
2.5. DNA Synthesis
2.6. Migration Assay
2.7. Tube Formation Assay
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
3.1. PI3K and mTOR Pathways are Involved in ATP-induced DNA Synthesis in VVECs
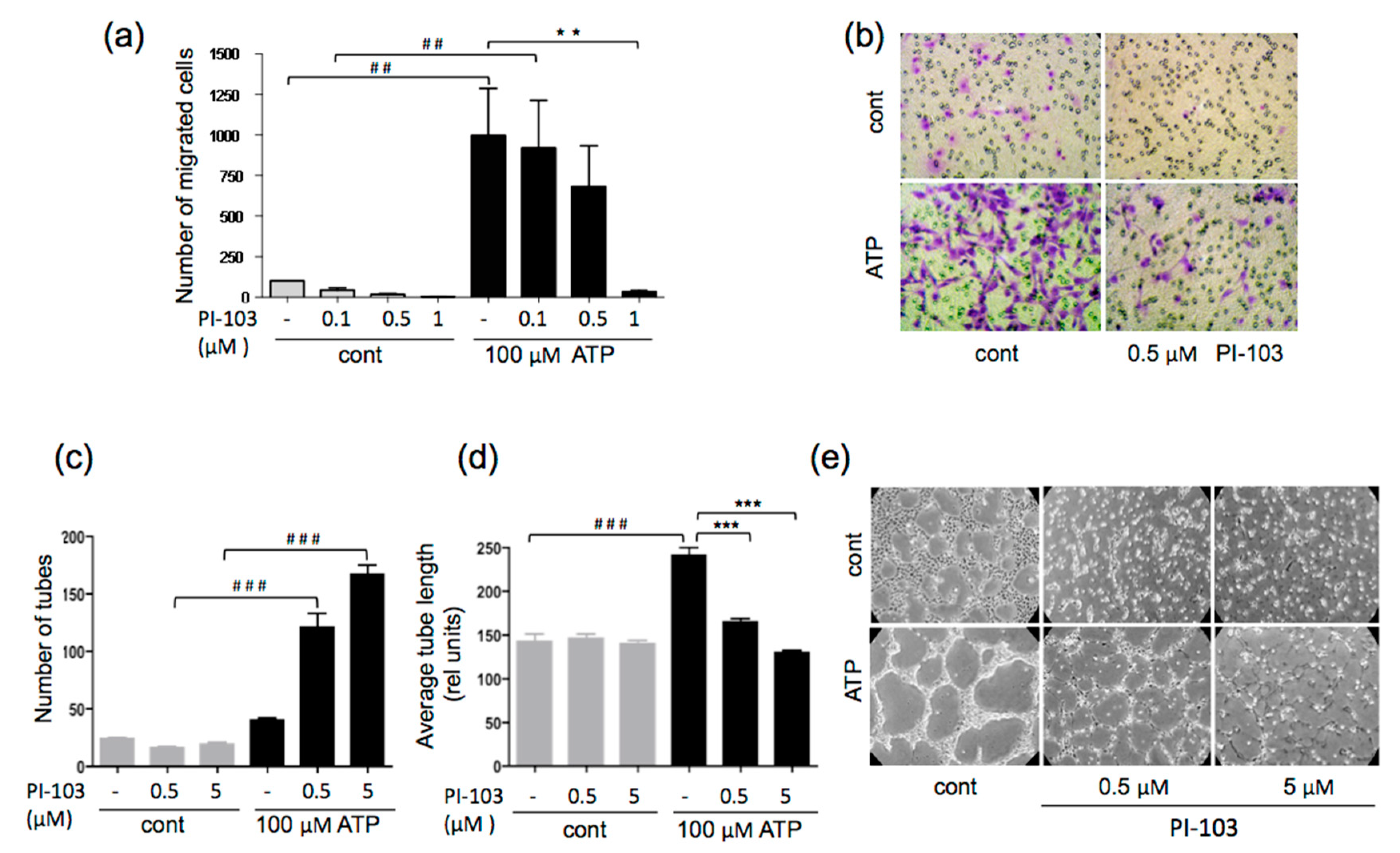
3.2. ATP-stimulated VVEC Migration and Tube Formation are PI3K- and mTOR-dependent
3.3. Transcription Factor Activation Profile Identifies Extracellular ATP-regulated TFs
3.4. ATP-mediated Phosphorylation of c-Jun, Foxo3a, and c-Myc is PI-103-dependent
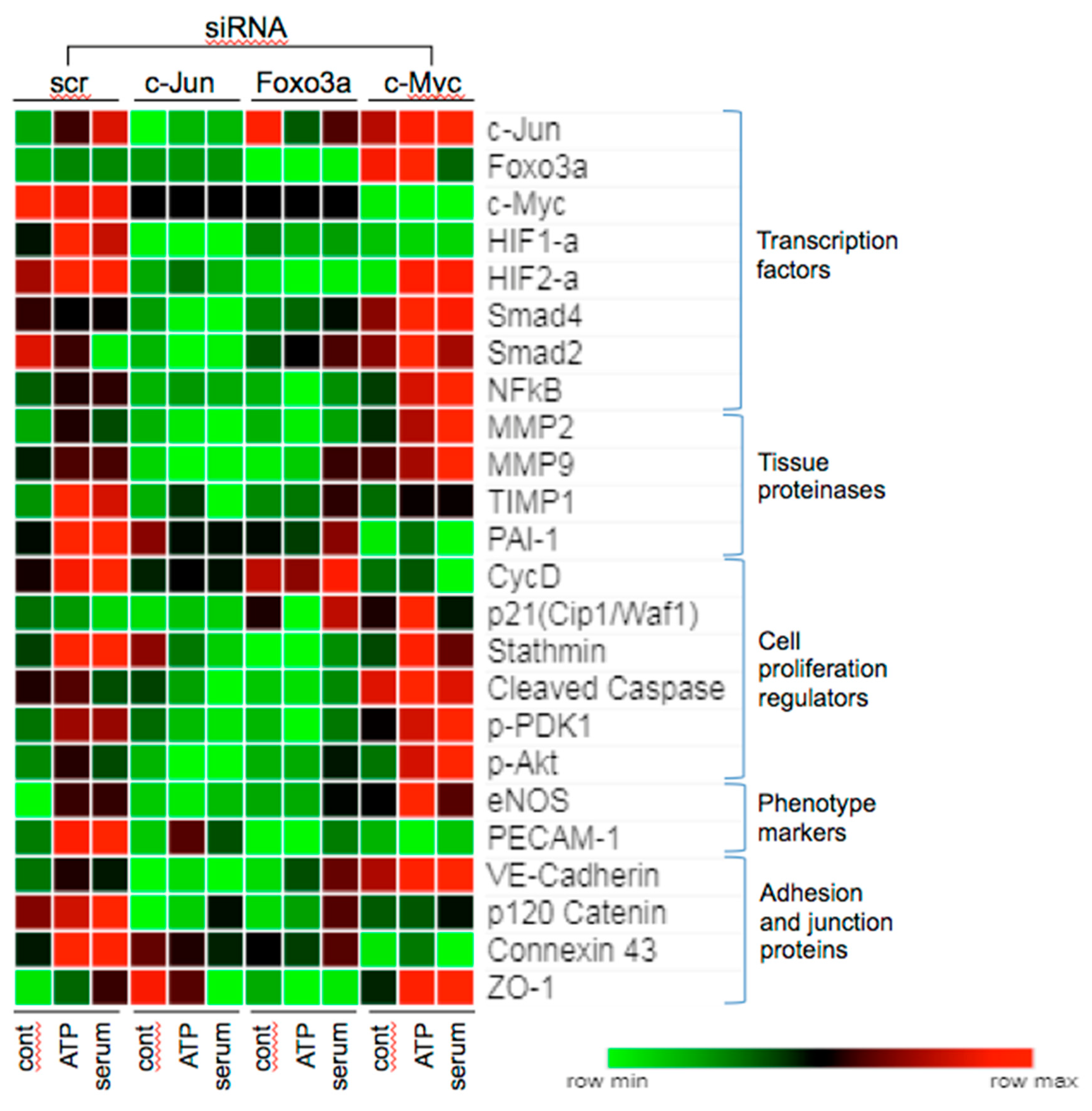
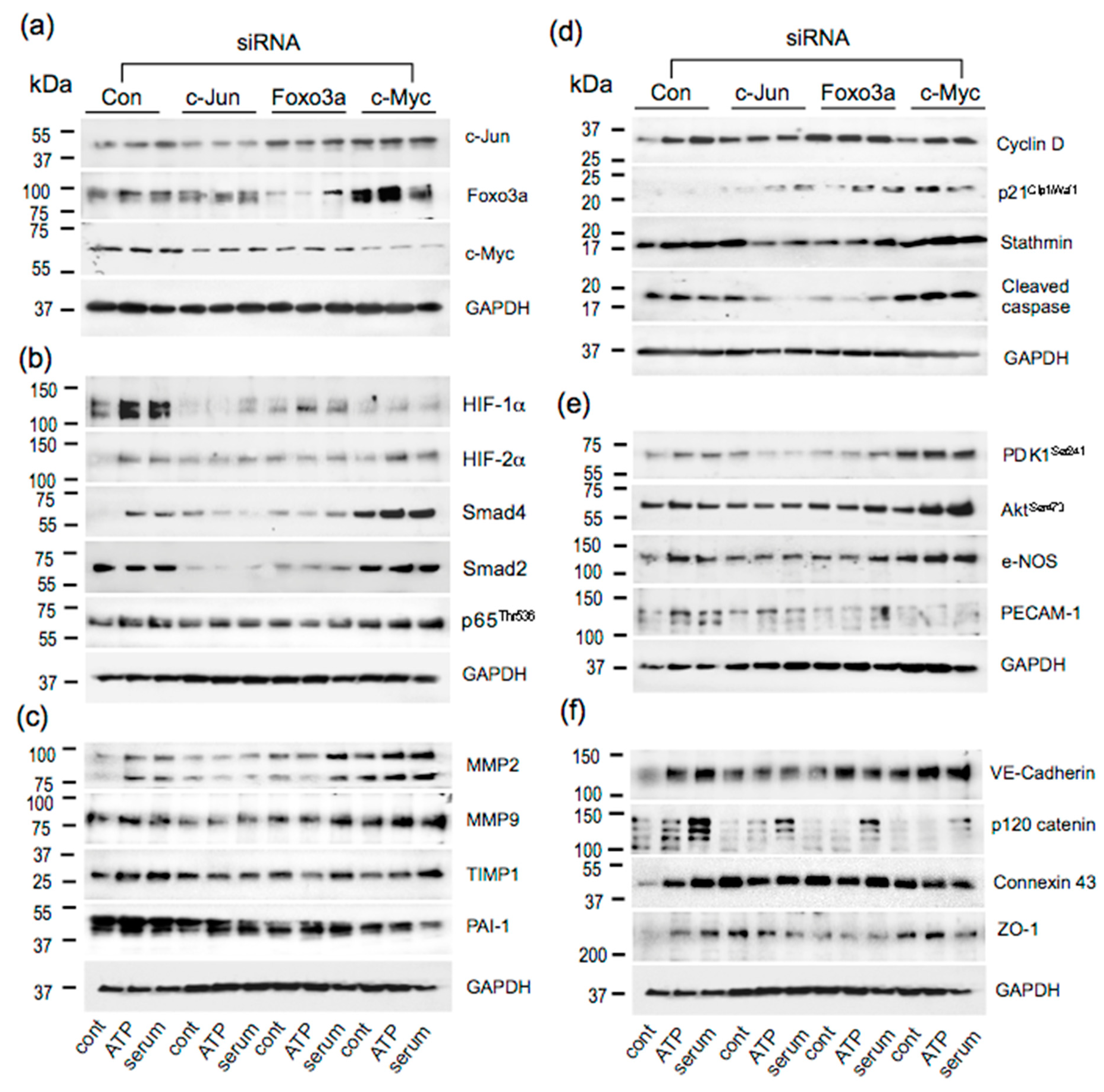
3.5. Effect of siRNA to c-Jun, Foxo3a, and c-Myc on Angiogenic Responses in VVEC
3.6. Effects of c-Jun, Foxo3a, and c-Myc KD on the Expression of Angiogenesis Associated Proteins
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Source | Catalog# | Dilution |
|---|---|---|---|
| p-AktSer473 | CST | CST4060 | 1:1000 |
| Akt | CST | CST9272 | 1:1000 |
| p-S6 | CST | CST2211 | 1:1000 |
| S6 | CST | CST2217 | 1:1000 |
| p-ERK1/2 | CST | CST4370 | 1:1000 |
| ERK | CST | CST9102 | 1:1000 |
| p-mTOR | CST | CST5536 | 1:1000 |
| mTOR | CST | CST2972 | 1:1000 |
| p-p70S6KThr421/Ser424 | CST | CST9204 | 1:1000 |
| p70S6K | CST | CST9202 | 1:1000 |
| p-PDKSer241 | CST | CST3061 | 1:1000 |
| Lamin AC | CST | CST477 | 1:6000 |
| c-JunSer73 | CST | CST8752 | 1:1000 |
| c-JunThr239 | Genetex | GTX50103 | 1:1000 |
| c-Jun | CST | CST9165 | 1:1000 |
| FoxO3aSer253 | CST | CST13129 | 1:1000 |
| FoxO3a | CST | CST12829 | 1:1000 |
| MycSer62 | CST | CST13748 | 1:1000 |
| Myc | CST | CST9402 | 1:1000 |
| HIF-1α | Novus | NB100654 | 1:1000 |
| HIF-2 | Novus | NB100132 | 1:1000 |
| Smad2 | CST | CST 5339 | 1:1000 |
| Smad4 | CST | CST4653 | 1:1000 |
| p65Thr536 | CST | CST4887 | 1:1000 |
| MMP2 | CST | CST4022 | 1:1000 |
| TIMP1 | CST | CST8946 | 1:1000 |
| MMP9 | CST | CST27647 | 1:1000 |
| PAI-1 | CST | CST11907 | 1:1000 |
| Cyclin D1 | SCBT | SC-753 | 1:1000 |
| Stathmin | Abcam | CST3352 | 1:1000 |
| PECAM-1 | CST | NBP171663 | 1:1000 |
| p21Cip1/Waf1 | CST | CST2947 | 1:1000 |
| Cleaved caspase3 | CST | CST9664 | 1:500 |
| PDK1 | CST | CST3062 | 1:1000 |
| e-NOS | CST | CST32027 | 1:1000 |
| VE-Cadherin | CST | CST2158 | 1:1000 |
| Connexin 43 | CST | CST3512 | 1:1000 |
| p120 Catenin | CST | CST4989 | 1:1000 |
| ZO-1 | CST | CST5406 | 1:1000 |
| GAPDH | CST | CST2118 | 1:5000 |
References
- Mulligan-Kehoe, M.J.; Simons, M. Vasa vasorum in normal and diseased arteries. Circulation 2014, 129, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Ritman, E.L.; Lerman, A. The dynamic vasa vasorum. Cardiovasc Res. 2007, 75, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, K.R.; Yeager, M.E.; El Kasmi, K.C.; Nozik-Grayck, E.; Gerasimovskaya, E.V.; Li, M.; Riddle, S.R.; Frid, M.G. The adventitia: Essential regulator of vascular wall structure and function. Annu Rev. Physiol 2013, 75, 23–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulligan-Kehoe, M.J. The vasa vasorum in diseased and nondiseased arteries. Am. J. Physiol Heart Circ. Physiol 2010, 298, H295–H305. [Google Scholar] [CrossRef] [Green Version]
- Kuwahara, F.; Kai, H.; Tokuda, K.; Shibata, R.; Kusaba, K.; Tahara, N.; Niiyama, H.; Nagata, T.; Imaizumi, T. Hypoxia-inducible factor-1alpha/vascular endothelial growth factor pathway for adventitial vasa vasorum formation in hypertensive rat aorta. Hypertension 2002, 39, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.C.; Sedding, D.G.; Haverich, A. Targeting vasa vasorum dysfunction to prevent atherosclerosis. Vascular pharmacology 2017, 96–98, 5–10. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: A malignant transformation. Cardiovasc Diabetol 2004, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Davie, N.J.; Crossno, J.T., Jr.; Frid, M.G.; Hofmeister, S.E.; Reeves, J.T.; Hyde, D.M.; Carpenter, T.C.; Brunetti, J.A.; McNiece, I.K.; Stenmark, K.R. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: Contribution of progenitor cells. Am. J. Physiol Lung Cell Mol. Physiol 2004, 286, L668–L678. [Google Scholar] [CrossRef]
- Stenmark, K.R.; Nozik-Grayck, E.; Gerasimovskaya, E.; Anwar, A.; Li, M.; Riddle, S.; Frid, M. The adventitia: Essential role in pulmonary vascular remodeling. Compr Physiol 2011, 1, 141–161. [Google Scholar] [CrossRef] [Green Version]
- Montani, D.; Perros, F.; Gambaryan, N.; Girerd, B.; Dorfmuller, P.; Price, L.C.; Huertas, A.; Hammad, H.; Lambrecht, B.; Simonneau, G.; et al. C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am. J. Respir Crit Care Med. 2011, 184, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purine and pyrimidine receptors. Cell Mol. Life Sci 2007, 64, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef] [PubMed]
- Lyubchenko, T.; Woodward, H.; Veo, K.D.; Burns, N.; Nijmeh, H.; Liubchenko, G.A.; Stenmark, K.R.; Gerasimovskaya, E.V. P2Y1 and P2Y13 purinergic receptors mediate Ca2+ signaling and proliferative responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol Cell Physiol 2011, 300, C266–C275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [Green Version]
- Bours, M.J.; Swennen, E.L.; Di Virgilio, F.; Cronstein, B.N.; Dagnelie, P.C. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther 2006, 112, 358–404. [Google Scholar] [CrossRef]
- Burnstock, G. Pathophysiology and therapeutic potential of purinergic signaling. Pharmacol Rev. 2006, 58, 58–86. [Google Scholar] [CrossRef]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Woodward, H.N.; Anwar, A.; Riddle, S.; Taraseviciene-Stewart, L.; Fragoso, M.; Stenmark, K.R.; Gerasimovskaya, E.V. PI3K, Rho, and ROCK play a key role in hypoxia-induced ATP release and ATP-stimulated angiogenic responses in pulmonary artery vasa vasorum endothelial cells. Am. J. Physiol Lung Cell Mol. Physiol 2009, 297, L954–L964. [Google Scholar] [CrossRef] [Green Version]
- Gerasimovskaya, E.V.; Ahmad, S.; White, C.W.; Jones, P.L.; Carpenter, T.C.; Stenmark, K.R. Extracellular ATP is an autocrine/paracrine regulator of hypoxia-induced adventitial fibroblast growth. Signaling through extracellular signal-regulated kinase-1/2 and the Egr-1 transcription factor. J. Biol Chem 2002, 277, 44638–44650. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid, M.R.; Guo, S.; Minami, T.; Spokes, K.C.; Ueki, K.; Skurk, C.; Walsh, K.; Aird, W.C. Vascular endothelial growth factor activates PI3K/Akt/forkhead signaling in endothelial cells. Arterioscler Thromb Vasc Biol 2004, 24, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Skurk, C.; Maatz, H.; Kim, H.S.; Yang, J.; Abid, M.R.; Aird, W.C.; Walsh, K. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol Chem 2004, 279, 1513–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerasimovskaya, E.V.; Woodward, H.N.; Tucker, D.A.; Stenmark, K.R. Extracellular ATP is a pro-angiogenic factor for pulmonary artery vasa vasorum endothelial cells. Angiogenesis 2008, 11, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, H.W.; Hernandez-Rodriguez, B.; Kim, J.; Kim, K.P.; Enriquez-Gasca, R.; Yoon, J.; Adams, S.; Scholer, H.R.; Vaquerizas, J.M.; Adams, R.H. Transcriptional regulation of endothelial cell behavior during sprouting angiogenesis. Nat. Commun 2017, 8, 726. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ. Res. 2002, 90, 1243–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Val, S.; Black, B.L. Transcriptional control of endothelial cell development. Dev. Cell 2009, 16, 180–195. [Google Scholar] [CrossRef] [Green Version]
- Oettgen, P. Transcriptional regulation of vascular development. Circ. Res. 2001, 89, 380–388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.C.; Young, D.A.; Waters, J.G.; Rowan, A.D.; Chantry, A.; Edwards, D.R.; Clark, I.M. The comparative role of activator protein 1 and Smad factors in the regulation of Timp-1 and MMP-1 gene expression by transforming growth factor-beta 1. J. Biol Chem 2003, 278, 10304–10313. [Google Scholar] [CrossRef] [Green Version]
- Hamik, A.; Wang, B.; Jain, M.K. Transcriptional regulators of angiogenesis. Arterioscler Thromb Vasc Biol 2006, 26, 1936–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharjee, S.; Renganaath, K.; Mehrotra, R.; Mehrotra, S. Combinatorial control of gene expression. Biomed. Res. Int 2013, 2013, 407263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, M.K.; Su, Y.; Yeung, W.W.; Wong, Y.H. Regulation of transcription factors by heterotrimeric G proteins. Curr Mol. Pharmacol 2009, 2, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Chapuis, N.; Bardet, V.; Tamburini, J.; Gallay, N.; Willems, L.; Knight, Z.A.; Shokat, K.M.; Azar, N.; Viguie, F.; et al. PI-103, a dual inhibitor of Class IA phosphatidylinositide 3-kinase and mTOR, has antileukemic activity in AML. Leukemia 2008, 22, 1698–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raynaud, F.I.; Eccles, S.; Clarke, P.A.; Hayes, A.; Nutley, B.; Alix, S.; Henley, A.; Di-Stefano, F.; Ahmad, Z.; Guillard, S.; et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007, 67, 5840–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.Y.; Luciano, A.K.; Ackah, E.; Rodriguez-Vita, J.; Bancroft, T.A.; Eichmann, A.; Simons, M.; Kyriakides, T.R.; Morales-Ruiz, M.; Sessa, W.C. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl Acad Sci U S A 2014, 111, 12865–12870. [Google Scholar] [CrossRef] [Green Version]
- Chou, C.K.; Lee, D.F.; Sun, H.L.; Li, L.Y.; Lin, C.Y.; Huang, W.C.; Hsu, J.M.; Kuo, H.P.; Yamaguchi, H.; Wang, Y.N.; et al. The suppression of MAD1 by AKT-mediated phosphorylation activates MAD1 target genes transcription. Mol. Carcinog 2009, 48, 1048–1058. [Google Scholar] [CrossRef] [Green Version]
- Tullai, J.W.; Graham, J.R.; Cooper, G.M. A GSK-3-mediated transcriptional network maintains repression of immediate early genes in quiescent cells. Cell Cycle 2011, 10, 3072–3077. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.Y.; Tang, F.; Yao, K.; Lu, C.; Zhu, F.; Zheng, D.; Pugliese, A.; Bode, A.M.; Dong, Z. Cyclin-dependent kinase-3-mediated c-Jun phosphorylation at Ser63 and Ser73 enhances cell transformation. Cancer Res. 2009, 69, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Hoeffler, W.K.; Levinson, A.D.; Bauer, E.A. Activation of c-Jun transcription factor by substitution of a charged residue in its N-terminal domain. Nucleic Acids Res. 1994, 22, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wei, B.; Liu, S.; Guo, C.; Wu, N.; Liu, Q.; Sun, M.Z. Anxa5 mediates the in vitro malignant behaviours of murine hepatocarcinoma Hca-F cells with high lymph node metastasis potential preferentially via ERK2/p-ERK2/c-Jun/p-c-Jun(Ser73) and E-cadherin. Biomed. Pharmacother 2016, 84, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.; Davis, R.J.; McLaren, A.; Cohen, P. A reinvestigation of the multisite phosphorylation of the transcription factor c-Jun. EMBO J. 2003, 22, 3876–3886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- van Hinsbergh, V.W.; Koolwijk, P. Endothelial sprouting and angiogenesis: Matrix metalloproteinases in the lead. Cardiovasc Res. 2008, 78, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Toricelli, M.; Melo, F.H.M.; Hunger, A.; Zanatta, D.; Strauss, B.E.; Jasiulionis, M.G. Timp1 Promotes Cell Survival by Activating the PDK1 Signaling Pathway in Melanoma. Cancers (Basel) 2017, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [Green Version]
- Kandasamy, K.; Escue, R.; Manna, J.; Adebiyi, A.; Parthasarathi, K. Changes in endothelial connexin 43 expression inversely correlate with microvessel permeability and VE-cadherin expression in endotoxin-challenged lungs. Am. J. Physiol Lung Cell Mol. Physiol 2015, 309, L584–L592. [Google Scholar] [CrossRef] [Green Version]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol 2015, 208, 821–838. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Feng, X.; Liu, F.; Zhang, P.; Liang, J.; Tang, X. Roles of PI3K/Akt and c-Jun signaling pathways in human papillomavirus type 16 oncoprotein-induced HIF-1alpha, VEGF, and IL-8 expression and in vitro angiogenesis in non-small cell lung cancer cells. PLoS One 2014, 9, e103440. [Google Scholar] [CrossRef]
- Singh, N.K.; Quyen, D.V.; Kundumani-Sridharan, V.; Brooks, P.C.; Rao, G.N. AP-1 (Fra-1/c-Jun)-mediated induction of expression of matrix metalloproteinase-2 is required for 15S-hydroxyeicosatetraenoic acid-induced angiogenesis. J. Biol Chem 2010, 285, 16830–16843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vleugel, M.M.; Greijer, A.E.; Bos, R.; van der Wall, E.; van Diest, P.J. c-Jun activation is associated with proliferation and angiogenesis in invasive breast cancer. Hum. Pathol 2006, 37, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Urbich, C.; Sasaki, K.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Invest. 2005, 115, 2382–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandramohan, V.; Mineva, N.D.; Burke, B.; Jeay, S.; Wu, M.; Shen, J.; Yang, W.; Hann, S.R.; Sonenshein, G.E. c-Myc represses FOXO3a-mediated transcription of the gene encoding the p27(Kip1) cyclin dependent kinase inhibitor. Journal of cellular biochemistry 2008, 104, 2091–2106. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol 2015, 27, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Dejana, E.; Taddei, A.; Randi, A.M. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta 2007, 1775, 298–312. [Google Scholar] [CrossRef]
- Knies-Bamforth, U.E.; Fox, S.B.; Poulsom, R.; Evan, G.I.; Harris, A.L. c-Myc interacts with hypoxia to induce angiogenesis in vivo by a vascular endothelial growth factor-dependent mechanism. Cancer Res. 2004, 64, 6563–6570. [Google Scholar] [CrossRef] [Green Version]
- Skurk, C.; Maatz, H.; Rocnik, E.; Bialik, A.; Force, T.; Walsh, K. Glycogen-Synthase Kinase3beta/beta-catenin axis promotes angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ. Res. 2005, 96, 308–318. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.D.; Hackett, S.F.; Hirota, K.; Oshima, Y.; Cai, Z.; Berg-Dixon, S.; Rowan, A.; Yan, Z.; Campochiaro, P.A.; Semenza, G.L. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ. Res. 2003, 93, 1074–1081. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhang, Y.P.; Kirsner, R.S. Angiogenesis in wound repair: Angiogenic growth factors and the extracellular matrix. Microsc Res. Tech. 2003, 60, 107–114. [Google Scholar] [CrossRef]
- Sluimer, J.C.; Daemen, M.J. Novel concepts in atherogenesis: Angiogenesis and hypoxia in atherosclerosis. J. Pathol 2009, 218, 7–29. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis 2008, 11, 121–140. [Google Scholar] [CrossRef]
- Gerasimovskaya, E.V.; Tucker, D.A.; Weiser-Evans, M.; Wenzlau, J.M.; Klemm, D.J.; Banks, M.; Stenmark, K.R. Extracellular ATP-induced proliferation of adventitial fibroblasts requires phosphoinositide 3-kinase, Akt, mammalian target of rapamycin, and p70 S6 kinase signaling pathways. J. Biol Chem 2005, 280, 1838–1848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullamsetti, S.S.; Perros, F.; Chelladurai, P.; Yuan, J.; Stenmark, K. Transcription factors, transcriptional coregulators, and epigenetic modulation in the control of pulmonary vascular cell phenotype: Therapeutic implications for pulmonary hypertension (2015 Grover Conference series). Pulm Circ. 2016, 6, 448–464. [Google Scholar] [CrossRef] [Green Version]
- Ennis, B.W.; Fultz, K.E.; Smith, K.A.; Westwick, J.K.; Zhu, D.; Boluro-Ajayi, M.; Bilter, G.K.; Stein, B. Inhibition of tumor growth, angiogenesis, and tumor cell proliferation by a small molecule inhibitor of c-Jun N-terminal kinase. J. Pharmacol Exp. Ther 2005, 313, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Dass, C.R.; Sumithran, E.; Di Girolamo, N.; Sun, L.Q.; Khachigian, L.M. Effect of deoxyribozymes targeting c-Jun on solid tumor growth and angiogenesis in rodents. J. Natl Cancer Inst. 2004, 96, 683–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, R.G.; Waldman, A.; Zhang, G.; Mitchell, A.; Tedla, N.; Cai, H.; Geczy, C.R.; Chesterman, C.N.; Perry, M.; Khachigian, L.M. Suppression of vascular permeability and inflammation by targeting of the transcription factor c-Jun. Nat. Biotechnol 2006, 24, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cai, S.; Wang, G.; Cao, X.; Yang, X.; Luo, X.; Feng, Y.; Hu, J. c-Myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1alpha. Biochem Biophys Res. Commun 2013, 430, 505–511. [Google Scholar] [CrossRef]
- Song, Y.; Wu, J.; Oyesanya, R.A.; Lee, Z.; Mukherjee, A.; Fang, X. Sp-1 and c-Myc mediate lysophosphatidic acid-induced expression of vascular endothelial growth factor in ovarian cancer cells via a hypoxia-inducible factor-1-independent mechanism. Clin. Cancer Res. 2009, 15, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244, 228p following 244. [Google Scholar] [CrossRef]
- Schwarte-Waldhoff, I.; Schmiegel, W. Smad4 transcriptional pathways and angiogenesis. Int J. Gastrointest Cancer 2002, 31, 47–59. [Google Scholar] [CrossRef]
- Zakrzewicz, A.; Kouri, F.M.; Nejman, B.; Kwapiszewska, G.; Hecker, M.; Sandu, R.; Dony, E.; Seeger, W.; Schermuly, R.T.; Eickelberg, O.; et al. The transforming growth factor-beta/Smad2,3 signalling axis is impaired in experimental pulmonary hypertension. Eur Respir J. 2007, 29, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Florea, V.; Bhagavatula, N.; Simovic, G.; Macedo, F.Y.; Fock, R.A.; Rodrigues, C.O. c-Myc is essential to prevent endothelial pro-inflammatory senescent phenotype. PLoS One 2013, 8, e73146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magid, R.; Murphy, T.J.; Galis, Z.S. Expression of matrix metalloproteinase-9 in endothelial cells is differentially regulated by shear stress. Role of c-Myc. J. Biol Chem 2003, 278, 32994–32999. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.; Ferber, E.C.; Schulze, A. Antagonism between FOXO and MYC Regulates Cellular Powerhouse. Front. Oncol 2013, 3, 96. [Google Scholar] [CrossRef] [Green Version]
- Amente, S.; Zhang, J.; Lavadera, M.L.; Lania, L.; Avvedimento, E.V.; Majello, B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011, 39, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, C.; Marquardt, J.; Bras, A.; Medema, R.H.; Eilers, M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004, 23, 2830–2840. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Zhang, L.; Han, W.; Shen, T.; Ma, C.; Liu, Y.; Nie, X.; Liu, M.; Ran, Y.; Zhu, D. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. J. Lipid Res. 2012, 53, 1093–1105. [Google Scholar] [CrossRef] [Green Version]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.H.; Liu, L.Z. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets 2008, 8, 19–26. [Google Scholar] [CrossRef]
- Balsara, R.D.; Castellino, F.J.; Ploplis, V.A. A novel function of plasminogen activator inhibitor-1 in modulation of the AKT pathway in wild-type and plasminogen activator inhibitor-1-deficient endothelial cells. J. Biol Chem 2006, 281, 22527–22536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollmark, J.; Ravi, S.; Sun, B.; Shipman, S.; Buitendijk, M.; Simons, M.; Mulligan-Kehoe, M.J. Antiangiogenic activity of rPAI-1(23) promotes vasa vasorum regression in hypercholesterolemic mice through a plasmin-dependent mechanism. Circ. Res. 2011, 108, 1419–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebbagh, M.; Renvoize, C.; Hamelin, J.; Riche, N.; Bertoglio, J.; Breard, J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat. Cell Biol 2001, 3, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Nhan, T.Q.; Liles, W.C.; Schwartz, S.M. Physiological functions of caspases beyond cell death. Am. J. Pathol 2006, 169, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol Chem 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strassheim, D.; Karoor, V.; Nijmeh, H.; Weston, P.; Lapel, M.; Schaack, J.; Sullivan, T.; Dempsey, E.C.; Stenmark, K.R.; Gerasimovskaya, E. c-Jun, Foxo3a, and c-Myc Transcription Factors are Key Regulators of ATP-Mediated Angiogenic Responses in Pulmonary Artery Vasa Vasorum Endothelial Cells †. Cells 2020, 9, 416. https://doi.org/10.3390/cells9020416
Strassheim D, Karoor V, Nijmeh H, Weston P, Lapel M, Schaack J, Sullivan T, Dempsey EC, Stenmark KR, Gerasimovskaya E. c-Jun, Foxo3a, and c-Myc Transcription Factors are Key Regulators of ATP-Mediated Angiogenic Responses in Pulmonary Artery Vasa Vasorum Endothelial Cells †. Cells. 2020; 9(2):416. https://doi.org/10.3390/cells9020416
Chicago/Turabian StyleStrassheim, Derek, Vijaya Karoor, Hala Nijmeh, Philip Weston, Martin Lapel, Jerome Schaack, Timothy Sullivan, Edward C. Dempsey, Kurt R. Stenmark, and Evgenia Gerasimovskaya. 2020. "c-Jun, Foxo3a, and c-Myc Transcription Factors are Key Regulators of ATP-Mediated Angiogenic Responses in Pulmonary Artery Vasa Vasorum Endothelial Cells †" Cells 9, no. 2: 416. https://doi.org/10.3390/cells9020416
APA StyleStrassheim, D., Karoor, V., Nijmeh, H., Weston, P., Lapel, M., Schaack, J., Sullivan, T., Dempsey, E. C., Stenmark, K. R., & Gerasimovskaya, E. (2020). c-Jun, Foxo3a, and c-Myc Transcription Factors are Key Regulators of ATP-Mediated Angiogenic Responses in Pulmonary Artery Vasa Vasorum Endothelial Cells †. Cells, 9(2), 416. https://doi.org/10.3390/cells9020416