Carbohydrate and Amino Acid Metabolism as Hallmarks for Innate Immune Cell Activation and Function



Abstract
:1. Carbohydrate and Amino Acid Metabolism
1.1. Carbohydrate Metabolism
1.2. Amino Acid Metabolism
2. Regulation of Carbohydrate and Amino Acid Metabolism in Innate Immunity
2.1. Macrophages—M1 Macrophages
2.2. Macrophages—M2 Macrophage
2.3. Dendritic Cells (DCs)
2.4. Neutrophils
2.5. Myeloid-Derived Suppressor Cells (MDSCs)
3. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig. 2003, 112, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.P.; Finlay, D.K. Glucose, glycolysis and lymphocyte responses. Mol. Immunol. 2015, 68, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, A.; Liu, J.; Zhou, Y.; Oltvai, Z.N. Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 2010, 4, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Vivancos, P.; de Simone, A.; Kiddle, G.; Foyer, C.H. Glutathione—Linking cell proliferation to oxidative stress. Free Radic. Biol. Med. 2015, 89, 1154–1164. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Baardman, J.; Verberk, S.G.S.; Prange, K.H.M.; van Weeghel, M.; van der Velden, S.; Ryan, D.G.; Wüst, R.C.I.; Neele, A.E.; Speijer, D.; Denis, S.W.; et al. A Defective Pentose Phosphate Pathway Reduces Inflammatory Macrophage Responses during Hypercholesterolemia. Cell Rep. 2018, 25, 2044–2052.e5. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef]
- Infantino, V.; Iacobazzi, V.; Palmieri, F.; Menga, A. ATP-citrate lyase is essential for macrophage inflammatory response. Biochem. Biophys. Res. Commun. 2013, 440, 105–111. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A.J. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 2016, 167, 457–470.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, B.; Tannahill, G.M.; Murphy, M.P.; O’Neill, L.A.J. Metformin Inhibits the Production of Reactive Oxygen Species from NADH:Ubiquinone Oxidoreductase to Limit Induction of Interleukin-1β (IL-1β) and Boosts Interleukin-10 (IL-10) in Lipopolysaccharide (LPS)-activated Macrophages. J. Biol. Chem. 2015, 290, 20348–20359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikwana, V.M.; Khanna, M.; Baskaran, S.; Tagliabracci, V.S.; Contreras, C.J.; DePaoli-Roach, A.; Roach, P.J.; Hurley, T.D. Structural basis for 2′-phosphate incorporation into glycogen by glycogen synthase. Proc. Natl. Acad. Sci. USA 2013, 110, 20976–20981. [Google Scholar] [CrossRef] [Green Version]
- Adeva-Andany, M.M.; González-Lucán, M.; Donapetry-García, C.; Fernández-Fernández, C.; Ameneiros-Rodríguez, E. Glycogen metabolism in humans. BBA Clin. 2016, 5, 85–100. [Google Scholar] [CrossRef] [Green Version]
- Khowaja, A.; Choi, I.-Y.; Seaquist, E.R.; Öz, G. In vivo Magnetic Resonance Spectroscopy of cerebral glycogen metabolism in animals and humans. Metab. Brain Dis. 2015, 30, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.S.; Lee, Y.M.; Cheung, Y.Y.; McDermott, D.H.; Murphy, P.M.; De Ravin, S.S.; Mansfield, B.C.; Chou, J.Y. Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood 2010, 116, 2783–2792. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.M.; Karnovsky, M.L.; Karnovsky, M.J. Glycogen accumulation in polymorphonuclear leukocytes, and other intracellular alterations that occur during inflammation. J. Cell Biol. 1982, 95, 933–942. [Google Scholar] [CrossRef] [PubMed]
- De Giorgis, V.; Veggiotti, P. GLUT1 deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, J.; Rustad, P.I.; Kolnes, A.J.; Lai, Y.-C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2011, 2, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Zheng, X. The crystal structure of human UDP-glucose pyrophosphorylase reveals a latch effect that influences enzymatic activity. Biochem. J. 2012, 442, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Burwinkel, B.; Bakker, H.D.; Herschkovitz, E.; Moses, S.W.; Shin, Y.S.; Kilimann, M.W. Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI. Am. J. Hum. Genet. 1998, 62, 785–791. [Google Scholar] [CrossRef] [Green Version]
- Wisselaar, H.A.; Kroos, M.A.; Hermans, M.M.; van Beeumen, J.; Reuser, A.J. Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation. J. Biol. Chem. 1993, 268, 2223–2231. [Google Scholar]
- Jun, H.S.; Weinstein, D.A.; Lee, Y.M.; Mansfield, B.C.; Chou, J.Y. Molecular mechanisms of neutrophil dysfunction in glycogen storage disease type Ib. Blood 2014, 123, 2843–2853. [Google Scholar] [CrossRef] [Green Version]
- Melis, D.; Della Casa, R.; Balivo, F.; Minopoli, G.; Rossi, A.; Salerno, M.; Andria, G.; Parenti, G. Involvement of endocrine system in a patient affected by glycogen storage disease 1b: Speculation on the role of autoimmunity. Ital. J. Pediatr. 2014, 40, 30. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.M. Glucose transport families SLC5 and SLC50. Mol. Asp. Med. 2013, 34, 183–196. [Google Scholar] [CrossRef]
- Frey, P.A. The Leloir pathway: A mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996, 10, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Coelho, A.I.; Berry, G.T.; Rubio-Gozalbo, M.E. Galactose metabolism and health. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Vuckovic, I.; Jeon, R.; Lerman, A.; Folmes, C.D.; Dzeja, P.P.; Herrmann, J. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coss, K.P.; Byrne, J.C.; Coman, D.J.; Adamczyk, B.; Abrahams, J.L.; Saldova, R.; Brown, A.Y.; Walsh, O.; Hendroff, U.; Carolan, C.; et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol. Genet. Metab. 2012, 105, 212–220. [Google Scholar] [CrossRef]
- Coss, K.P.; Hawkes, C.P.; Adamczyk, B.; Stöckmann, H.; Crushell, E.; Saldova, R.; Knerr, I.; Rubio-Gozalbo, M.E.; Monavari, A.A.; Rudd, P.M.; et al. N-glycan abnormalities in children with galactosemia. J. Proteome Res. 2014, 13, 385–394. [Google Scholar] [CrossRef]
- Taskinen, M.-R.; Söderlund, S.; Bogl, L.H.; Hakkarainen, A.; Matikainen, N.; Pietiläinen, K.H.; Räsänen, S.; Lundbom, N.; Björnson, E.; Eliasson, B.; et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J. Intern. Med. 2017, 282, 187–201. [Google Scholar] [CrossRef]
- Mirtschink, P.; Jang, C.; Arany, Z.; Krek, W. Fructose metabolism, cardiometabolic risk, and the epidemic of coronary artery disease. Eur. Heart J. 2018, 39, 2497–2505. [Google Scholar] [CrossRef]
- Patel, C.; Douard, V.; Yu, S.; Gao, N.; Ferraris, R.P. Transport, metabolism, and endosomal trafficking-dependent regulation of intestinal fructose absorption. FASEB J. 2015, 29, 4046–4058. [Google Scholar] [CrossRef] [Green Version]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef]
- Rutledge, A.C.; Adeli, K. Fructose and the metabolic syndrome: Pathophysiology and molecular mechanisms. Nutr. Rev. 2007, 65, S13–S23. [Google Scholar] [CrossRef]
- Ferrante, A.W. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Diehl, A.M. Role of inflammation in nonalcoholic steatohepatitis. Curr. Opin. Gastroenterol. 2005, 21, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, N.; Agrawal, S.; Agrawal, A. High fructose-induced metabolic changes enhance inflammation in human dendritic cells. Clin. Exp. Immunol. 2019, 197, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Wagnerberger, S.; Spruss, A.; Kanuri, G.; Volynets, V.; Stahl, C.; Bischoff, S.C.; Bergheim, I. Toll-like receptors 1-9 are elevated in livers with fructose-induced hepatic steatosis. Br. J. Nutr. 2012, 107, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Lemos, H.; Huang, L.; Prendergast, G.C.; Mellor, A.L. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat. Rev. Cancer 2019, 19, 162–175. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Kedia-Mehta, N.; Finlay, D.K. Competition for nutrients and its role in controlling immune responses. Nat Commun. 2019, 10, 2123. [Google Scholar] [CrossRef]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for L-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef]
- Hibbs, J.B.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef]
- Marletta, M.A.; Yoon, P.S.; Iyengar, R.; Leaf, C.D.; Wishnok, J.S. Macrophage oxidation of L-arginine to nitrite and nitrate: Nitric oxide is an intermediate. Biochemistry 1988, 27, 8706–8711. [Google Scholar] [CrossRef]
- Palmer, R.M.; Ashton, D.S.; Moncada, S. Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 1988, 333, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Davis, T.A.; Kim, S.W.; Li, P.; Rhoads, J.M.; Satterfield, M.C.; Smith, S.B.; Spencer, T.E.; Yin, Y. Arginine metabolism and nutrition in growth, health and disease. Amino Acids 2009, 37, 153–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, S.; Petrack, B. The mechanism of arginine synthesis from citrulline in kidney. J. Biol. Chem. 1953, 200, 175–185. [Google Scholar] [PubMed]
- Shima, Y.; Maeda, T.; Aizawa, S.; Tsuboi, I.; Kobayashi, D.; Kato, R.; Tamai, I. L-arginine import via cationic amino acid transporter CAT1 is essential for both differentiation and proliferation of erythrocytes. Blood 2006, 107, 1352–1356. [Google Scholar] [CrossRef] [Green Version]
- Tomi, M.; Kitade, N.; Hirose, S.; Yokota, N.; Akanuma, S.-I.; Tachikawa, M.; Hosoya, K. Cationic amino acid transporter 1-mediated L-arginine transport at the inner blood-retinal barrier. J. Neurochem. 2009, 111, 716–725. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.; Xie, Q.W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Rath, M.; Müller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [Green Version]
- Curi, R.; Lagranha, C.J.; Doi, S.Q.; Sellitti, D.F.; Procopio, J.; Pithon-Curi, T.C.; Corless, M.; Newsholme, P. Molecular mechanisms of glutamine action. J. Cell. Physiol. 2005, 204, 392–401. [Google Scholar] [CrossRef]
- Hensley, C.T.; Wasti, A.T.; DeBerardinis, R.J. Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Investig. 2013, 123, 3678–3684. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Cao, X. Epigenetic regulation of the innate immune response to infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Keast, D. Glutamine and macrophage function. Metab. Clin. Exp. 1992, 41, 1016–1020. [Google Scholar] [CrossRef]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Liu, P.-S.; Wang, H.; Li, X.; Chao, T.; Teav, T.; Christen, S.; Di Conza, G.; Cheng, W.-C.; Chou, C.-H.; Vavakova, M.; et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 2017, 18, 985–994. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.-C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Carr, E.L.; Kelman, A.; Wu, G.S.; Gopaul, R.; Senkevitch, E.; Aghvanyan, A.; Turay, A.M.; Frauwirth, K.A. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 2010, 185, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Crawford, J.; Cohen, H.J. The essential role of L-glutamine in lymphocyte differentiation in vitro. J. Cell. Physiol. 1985, 124, 275–282. [Google Scholar] [CrossRef]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Ron-Harel, N.; Notarangelo, G.; Ghergurovich, J.M.; Paulo, J.A.; Sage, P.T.; Santos, D.; Satterstrom, F.K.; Gygi, S.P.; Rabinowitz, J.D.; Sharpe, A.H.; et al. Defective respiration and one-carbon metabolism contribute to impaired naïve T cell activation in aged mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13347–13352. [Google Scholar] [CrossRef] [Green Version]
- Reid, M.A.; Allen, A.E.; Liu, S.; Liberti, M.V.; Liu, P.; Liu, X.; Dai, Z.; Gao, X.; Wang, Q.; Liu, Y.; et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar]
- Oxenkrug, G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol. Neurobiol. 2013, 48, 294–301. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, Y. Metabolic aspects of neuronal degeneration: From a NAD+ point of view. Neurosci. Res. 2019, 139, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Rodríguez-Prados, J.-C.; Través, P.G.; Cuenca, J.; Rico, D.; Aragonés, J.; Martín-Sanz, P.; Cascante, M.; Boscá, L. Substrate fate in activated macrophages: A comparison between innate, classic, and alternative activation. J. Immunol. 2010, 185, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Newsholme, P.; Newsholme, E.A. Rates of utilization of glucose, glutamine and oleate and formation of end-products by mouse peritoneal macrophages in culture. Biochem. J. 1989, 261, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Pavlou, S.; Wang, L.; Xu, H.; Chen, M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J. Inflamm. (Lond.) 2017, 14, 4. [Google Scholar] [CrossRef] [Green Version]
- Michl, J.; Ohlbaum, D.J.; Silverstein, S.C. 2-Deoxyglucose selectively inhibits Fc and complement receptor-mediated phagocytosis in mouse peritoneal macrophages II. Dissociation of the inhibitory effects of 2-deoxyglucose on phagocytosis and ATP generation. J. Exp. Med. 1976, 144, 1484–1493. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Pore, N.; Behrooz, A.; Ismail-Beigi, F.; Maity, A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J. Biol. Chem. 2001, 276, 9519–9525. [Google Scholar] [CrossRef] [Green Version]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L.; Jiang, B.H.; Leung, S.W.; Passantino, R.; Concordet, J.P.; Maire, P.; Giallongo, A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J. Biol. Chem. 1996, 271, 32529–32537. [Google Scholar] [CrossRef] [Green Version]
- Takeda, N.; O’Dea, E.L.; Doedens, A.; Kim, J.; Weidemann, A.; Stockmann, C.; Asagiri, M.; Simon, M.C.; Hoffmann, A.; Johnson, R.S. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010, 24, 491–501. [Google Scholar] [CrossRef] [Green Version]
- Peyssonnaux, C.; Datta, V.; Cramer, T.; Doedens, A.; Theodorakis, E.A.; Gallo, R.L.; Hurtado-Ziola, N.; Nizet, V.; Johnson, R.S. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J. Clin. Investig. 2005, 115, 1806–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Pearce, E.J. MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity 2017, 46, 730–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.-C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Nambu, A.; Jung, J.; Shibata, M.; Aksoylar, H.I.; Lei, J.; Xu, P.; Doan, M.T.; Jiang, H.; MacArthur, M.R.; et al. Glycerol phosphate shuttle enzyme GPD2 regulates macrophage inflammatory responses. Nat. Immunol. 2019, 20, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, M.; Furuyashiki, T.; Nakamura, T.; Kakutani, R.; Takata, H.; Ashida, H. Immunomodulatory activity of enzymatically synthesized glycogen and its digested metabolite in a co-culture system consisting of differentiated Caco-2 cells and RAW264.7 macrophages. Food Funct. 2013, 4, 1387–1393. [Google Scholar] [CrossRef]
- Kakutani, R.; Adachi, Y.; Takata, H.; Kuriki, T.; Ohno, N. Essential role of Toll-like receptor 2 in macrophage activation by glycogen. Glycobiology 2012, 22, 146–159. [Google Scholar] [CrossRef] [Green Version]
- Mitani, T.; Yoshioka, Y.; Furuyashiki, T.; Yamashita, Y.; Shirai, Y.; Ashida, H. Enzymatically synthesized glycogen inhibits colitis through decreasing oxidative stress. Free Radic. Biol. Med. 2017, 106, 355–367. [Google Scholar] [CrossRef]
- Kakutani, R.; Adachi, Y.; Kajiura, H.; Takata, H.; Kuriki, T.; Ohno, N. The effect of orally administered glycogen on anti-tumor activity and natural killer cell activity in mice. Int. Immunopharmacol. 2012, 12, 80–87. [Google Scholar] [CrossRef]
- Nussler, A.K.; Billiar, T.R.; Liu, Z.Z.; Morris, S.M. Coinduction of nitric oxide synthase and argininosuccinate synthetase in a murine macrophage cell line. Implications for regulation of nitric oxide production. J. Biol. Chem. 1994, 269, 1257–1261. [Google Scholar]
- Nagasaki, A.; Gotoh, T.; Takeya, M.; Yu, Y.; Takiguchi, M.; Matsuzaki, H.; Takatsuki, K.; Mori, M. Coinduction of nitric oxide synthase, argininosuccinate synthetase, and argininosuccinate lyase in lipopolysaccharide-treated rats. RNA blot, immunoblot, and immunohistochemical analyses. J. Biol. Chem. 1996, 271, 2658–2662. [Google Scholar] [CrossRef] [Green Version]
- Qualls, J.E.; Subramanian, C.; Rafi, W.; Smith, A.M.; Balouzian, L.; DeFreitas, A.A.; Shirey, K.A.; Reutterer, B.; Kernbauer, E.; Stockinger, S.; et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe 2012, 12, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Zhen, Y.; Ma, Z.; Li, H.; Yu, J.; Xu, Z.-G.; Wang, X.-Y.; Yi, H.; Yang, Y.-G. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci. Transl. Med. 2016, 8, 331ra40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, S.M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.; Saville, C.R.; Murray, P.J.; Cruickshank, S.M.; Hardman, M.J. Local arginase 1 activity is required for cutaneous wound healing. J. Investig. Dermatol. 2013, 133, 2461–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony, R.M.; Rutitzky, L.I.; Urban, J.F.; Stadecker, M.J.; Gause, W.C. Protective immune mechanisms in helminth infection. Nat. Rev. Immunol. 2007, 7, 975–987. [Google Scholar] [CrossRef] [Green Version]
- El Kasmi, K.C.; Qualls, J.E.; Pesce, J.T.; Smith, A.M.; Thompson, R.W.; Henao-Tamayo, M.; Basaraba, R.J.; König, T.; Schleicher, U.; Koo, M.-S.; et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 2008, 9, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Martín-Pérez, R.; Quinto, A.; Riera-Domingo, C.; De Tullio, G.; Hooper, D.C.; Lamers, W.H.; Ghesquière, B.; McVicar, D.W.; et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017, 20, 1654–1666. [Google Scholar] [CrossRef] [Green Version]
- Vannella, K.M.; Wynn, T.A. Mechanisms of Organ Injury and Repair by Macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Gause, W.C.; Wynn, T.A.; Allen, J.E. Type 2 immunity and wound healing: Evolutionary refinement of adaptive immunity by helminths. Nat. Rev. Immunol. 2013, 13, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.-C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J.; Snyder, N.W.; Worth, A.J.; Iyer, S.S.; Wang, J.; Ben-Sahra, I.; Byles, V.; Polynne-Stapornkul, T.; et al. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef] [PubMed]
- de-Brito, N.M.; Duncan-Moretti, J.; da-Costa, H.C.; Saldanha-Gama, R.; Paula-Neto, H.A.; Dorighello, G.G.; Simões, R.L.; Barja-Fidalgo, C. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118604. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; O’Neill, L.A.J. Mitochondria are the powerhouses of immunity. Nat. Immunol. 2017, 18, 488–498. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef]
- Murphy, M.P.; O’Neill, L.A.J. Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 2018, 174, 780–784. [Google Scholar] [CrossRef] [Green Version]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef]
- Peruzzotti-Jametti, L.; Bernstock, J.D.; Vicario, N.; Costa, A.S.H.; Kwok, C.K.; Leonardi, T.; Booty, L.M.; Bicci, I.; Balzarotti, B.; Volpe, G.; et al. Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 2018, 22, 355–368.e13. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.-Y.; Huang, T.-W.; Hsieh, Y.-T.; Wang, Y.-F.; Yen, C.-C.; Lee, G.-L.; Yeh, C.-C.; Peng, Y.-J.; Kuo, Y.-Y.; Wen, H.-T.; et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol. Cell 2020, 77, 213–227.e5. [Google Scholar] [CrossRef]
- Cordes, T.; Wallace, M.; Michelucci, A.; Divakaruni, A.S.; Sapcariu, S.C.; Sousa, C.; Koseki, H.; Cabrales, P.; Murphy, A.N.; Hiller, K.; et al. Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J. Biol. Chem. 2016, 291, 14274–14284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Davies, L.C.; Karwan, M.; Ileva, L.; Ozaki, M.K.; Cheng, R.Y.; Ridnour, L.A.; Annunziata, C.M.; Wink, D.A.; McVicar, D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 2018, 128, 3794–3805. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gut, P.; Verdin, E. The nexus of chromatin regulation and intermediary metabolism. Nature 2013, 502, 489–498. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1β Production. Cell Metab. 2019, 29, 1003–1011.e4. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160.e5. [Google Scholar] [CrossRef]
- Li, X.; Gong, W.; Wang, H.; Li, T.; Attri, K.S.; Lewis, R.E.; Kalil, A.C.; Bhinderwala, F.; Powers, R.; Yin, G.; et al. O-GlcNAc Transferase Suppresses Inflammation and Necroptosis by Targeting Receptor-Interacting Serine/Threonine-Protein Kinase 3. Immunity 2019, 50, 576–590.e6. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Menga, A.; Lebrun, A.; Hooper, D.C.; Butterfield, D.A.; Mazzone, M.; Castegna, A. Blockade of Glutamine Synthetase Enhances Inflammatory Response in Microglial Cells. Antioxid. Redox Signal. 2017, 26, 351–363. [Google Scholar] [CrossRef]
- Mazzone, M.; Menga, A.; Castegna, A. Metabolism and TAM functions-it takes two to tango. FEBS J. 2018, 285, 700–716. [Google Scholar] [CrossRef] [Green Version]
- Curti, A.; Trabanelli, S.; Salvestrini, V.; Baccarani, M.; Lemoli, R.M. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: Focus on hematology. Blood 2009, 113, 2394–2401. [Google Scholar] [CrossRef] [PubMed]
- Cronin, S.J.F.; Seehus, C.; Weidinger, A.; Talbot, S.; Reissig, S.; Seifert, M.; Pierson, Y.; McNeill, E.; Longhi, M.S.; Turnes, B.L.; et al. The metabolite BH4 controls T cell proliferation in autoimmunity and cancer. Nature 2018, 563, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Mondanelli, G.; Bianchi, R.; Pallotta, M.T.; Orabona, C.; Albini, E.; Iacono, A.; Belladonna, M.L.; Vacca, C.; Fallarino, F.; Macchiarulo, A.; et al. A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 2017, 46, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. (Berl.) 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Kutyavin, V.I.; Chawla, A. Tissue Immunometabolism: Development, Physiology, and Pathobiology. Cell Metab. 2017, 25, 11–26. [Google Scholar] [CrossRef]
- Nguyen-Lefebvre, A.T.; Horuzsko, A. Kupffer Cell Metabolism and Function. J. Enzymol. Metab. 2015, 1, 101. [Google Scholar]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Jager, J.; Aparicio-Vergara, M.; Aouadi, M. Liver innate immune cells and insulin resistance: The multiple facets of Kupffer cells. J. Intern. Med. 2016, 280, 209–220. [Google Scholar] [CrossRef] [Green Version]
- Tosello-Trampont, A.-C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.C.; Brown, B.D.; Shay, T.; Gautier, E.L.; Jojic, V.; Cohain, A.; Pandey, G.; Leboeuf, M.; Elpek, K.G.; Helft, J.; et al. Deciphering the transcriptional network of the dendritic cell lineage. Nat. Immunol. 2012, 13, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.J.; Everts, B. Dendritic cell metabolism. Nat. Rev. Immunol. 2015, 15, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinarich, F.; Duan, K.; Hamid, R.A.; Bijin, A.; Lin, W.X.; Poidinger, M.; Fairhurst, A.-M.; Connolly, J.E. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J. Immunol. 2015, 194, 5174–5186. [Google Scholar] [CrossRef] [Green Version]
- Jantsch, J.; Chakravortty, D.; Turza, N.; Prechtel, A.T.; Buchholz, B.; Gerlach, R.G.; Volke, M.; Gläsner, J.; Warnecke, C.; Wiesener, M.S.; et al. Hypoxia and Hypoxia-Inducible Factor-1α Modulate Lipopolysaccharide-Induced Dendritic Cell Activation and Function. J. Immunol. 2008, 180, 4697–4705. [Google Scholar] [CrossRef]
- Perrin-Cocon, L.; Aublin-Gex, A.; Diaz, O.; Ramière, C.; Peri, F.; André, P.; Lotteau, V. Toll-like Receptor 4-Induced Glycolytic Burst in Human Monocyte-Derived Dendritic Cells Results from p38-Dependent Stabilization of HIF-1α and Increased Hexokinase II Expression. J. Immunol. 2018, 201, 1510–1521. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.-C.; Smith, A.M.; Chang, C.-H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Manicassamy, S.; Tang, H.; Kasturi, S.P.; Pirani, A.; Murthy, N.; Pulendran, B. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat. Immunol. 2008, 9, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Amiel, E.; Everts, B.; Freitas, T.C.; King, I.L.; Curtis, J.D.; Pearce, E.L.; Pearce, E.J. Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J. Immunol. 2012, 189, 2151–2158. [Google Scholar] [CrossRef] [Green Version]
- Hussaarts, L.; Smits, H.H.; Schramm, G.; van der Ham, A.J.; van der Zon, G.C.; Haas, H.; Guigas, B.; Yazdanbakhsh, M. Rapamycin and omega-1: mTOR-dependent and -independent Th2 skewing by human dendritic cells. Immunol. Cell Biol. 2013, 91, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Bi, Y.; Xue, L.; Zhang, Y.; Yang, H.; Chen, X.; Lu, Y.; Zhang, Z.; Liu, H.; Wang, X.; et al. Dendritic cell SIRT1-HIF1α axis programs the differentiation of CD4+ T cells through IL-12 and TGF-β1. Proc. Natl. Acad. Sci. USA 2015, 112, E957–E965. [Google Scholar] [CrossRef] [Green Version]
- Guak, H.; Al Habyan, S.; Ma, E.H.; Aldossary, H.; Al-Masri, M.; Won, S.Y.; Ying, T.; Fixman, E.D.; Jones, R.G.; McCaffrey, L.M.; et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, X.; Chen, K.; Cheng, Y.; Liu, S.; Xia, M.; Chen, Y.; Zhu, H.; Li, Z.; Cao, X. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity 2019, 50, 600–615.e15. [Google Scholar] [CrossRef] [Green Version]
- Thwe, P.M.; Pelgrom, L.R.; Cooper, R.; Beauchamp, S.; Reisz, J.A.; D’Alessandro, A.; Everts, B.; Amiel, E. Cell-Intrinsic Glycogen Metabolism Supports Early Glycolytic Reprogramming Required for Dendritic Cell Immune Responses. Cell Metab. 2017, 26, 558–567.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.-H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajwa, G.; DeBerardinis, R.J.; Shao, B.; Hall, B.; Farrar, J.D.; Gill, M.A. Cutting Edge: Critical Role of Glycolysis in Human Plasmacytoid Dendritic Cell Antiviral Responses. J. Immunol. 2016, 196, 2004–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raychaudhuri, D.; Bhattacharya, R.; Sinha, B.P.; Liu, C.S.C.; Ghosh, A.R.; Rahaman, O.; Bandopadhyay, P.; Sarif, J.; D’Rozario, R.; Paul, S.; et al. Lactate Induces Pro-tumor Reprogramming in Intratumoral Plasmacytoid Dendritic Cells. Front. Immunol. 2019, 10, 1878. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic Control of Dendritic Cell Functions: Digesting Information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [Green Version]
- Tran Janco, J.M.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-infiltrating dendritic cells in cancer pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Sharma, M.D.; Hou, D.; Baban, B.; Lee, J.R.; Antonia, S.J.; Messina, J.L.; Chandler, P.; Koni, P.A.; Mellor, A.L. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Investig. 2004, 114, 280–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, C.; Sun, A.; Zheng, Y.; Wang, L.; Cao, X. Tumor-educated CD11bhighIalow regulatory dendritic cells suppress T cell response through arginase I. J. Immunol. 2009, 182, 6207–6216. [Google Scholar] [CrossRef] [Green Version]
- Narita, Y.; Kitamura, H.; Wakita, D.; Sumida, K.; Masuko, K.; Terada, S.; Nakano, K.; Nishimura, T. The key role of IL-6-arginase cascade for inducing dendritic cell-dependent CD4(+) T cell dysfunction in tumor-bearing mice. J. Immunol. 2013, 190, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Norian, L.A.; Rodriguez, P.C.; O’Mara, L.A.; Zabaleta, J.; Ochoa, A.C.; Cella, M.; Allen, P.M. Tumor-infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L-arginine metabolism. Cancer Res. 2009, 69, 3086–3094. [Google Scholar] [CrossRef] [Green Version]
- Gunzer, M. Traps and hyper inflammation—New ways that neutrophils promote or hinder survival. Br. J. Haematol. 2014, 164, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Soehnlein, O.; Steffens, S.; Hidalgo, A.; Weber, C. Neutrophils as protagonists and targets in chronic inflammation. Nat. Rev. Immunol. 2017, 17, 248–261. [Google Scholar] [CrossRef]
- Maianski, N.A.; Geissler, J.; Srinivasula, S.M.; Alnemri, E.S.; Roos, D.; Kuijpers, T.W. Functional characterization of mitochondria in neutrophils: A role restricted to apoptosis. Cell Death Differ. 2004, 11, 143–153. [Google Scholar] [CrossRef]
- Veiga-da-Cunha, M.; Chevalier, N.; Stephenne, X.; Defour, J.-P.; Paczia, N.; Ferster, A.; Achouri, Y.; Dewulf, J.P.; Linster, C.L.; Bommer, G.T.; et al. Failure to eliminate a phosphorylated glucose analog leads to neutropenia in patients with G6PT and G6PC3 deficiency. Proc. Natl. Acad. Sci. USA 2019, 116, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Boztug, K.; Appaswamy, G.; Ashikov, A.; Schäffer, A.A.; Salzer, U.; Diestelhorst, J.; Germeshausen, M.; Brandes, G.; Lee-Gossler, J.; Noyan, F.; et al. A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med. 2009, 360, 32–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amankulor, N.M.; Kim, Y.; Arora, S.; Kargl, J.; Szulzewsky, F.; Hanke, M.; Margineantu, D.H.; Rao, A.; Bolouri, H.; Delrow, J.; et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017, 31, 774–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Y.; Ledderose, C.; Graf, A.F.; Brix, B.; Birsak, T.; Lee, A.; Zhang, J.; Junger, W.G. mTOR and differential activation of mitochondria orchestrate neutrophil chemotaxis. J. Cell Biol. 2015, 210, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cao, L.; Jeffries, J.; Zhu, X.; Staiger, C.J.; Deng, Q. Neutrophil-specific knockout demonstrates a role for mitochondria in regulating neutrophil motility in zebrafish. Dis. Model. Mech. 2018, 11, dmm033027. [Google Scholar] [CrossRef] [Green Version]
- Sbarra, A.J.; Karnovsky, M.L. The biochemical basis of phagocytosis. I. Metabolic changes during the ingestion of particles by polymorphonuclear leukocytes. J. Biol. Chem. 1959, 234, 1355–1362. [Google Scholar]
- Borregaard, N.; Herlin, T. Energy metabolism of human neutrophils during phagocytosis. J. Clin. Investig. 1982, 70, 550–557. [Google Scholar] [CrossRef]
- Azevedo, E.P.; Rochael, N.C.; Guimarães-Costa, A.B.; de Souza-Vieira, T.S.; Ganilho, J.; Saraiva, E.M.; Palhano, F.L.; Foguel, D. A Metabolic Shift toward Pentose Phosphate Pathway Is Necessary for Amyloid Fibril-and Phorbol 12-Myristate 13-Acetate-induced Neutrophil Extracellular Trap (NET) Formation. J. Biol. Chem. 2015, 290, 22174–22183. [Google Scholar] [CrossRef] [Green Version]
- Cooper, M.R.; DeChatelet, L.R.; McCall, C.E.; LaVia, M.F.; Spurr, C.L.; Baehner, R.L. Complete deficiency of leukocyte glucose-6-phosphate dehydrogenase with defective bactericidal activity. J. Clin. Investig. 1972, 51, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Gray, G.R.; Stamatoyannopoulos, G.; Naiman, S.C.; Kliman, M.R.; Klebanoff, S.J.; Austin, T.; Yoshida, A.; Robinson, G.C. Neutrophil dysfunction, chronic granulomatous disease, and non-spherocytic haemolytic anaemia caused by complete deficiency of glucose-6-phosphate dehydrogenase. Lancet 1973, 2, 530–534. [Google Scholar] [CrossRef]
- Curi, T.C.; De Melo, M.P.; De Azevedo, R.B.; Zorn, T.M.; Curi, R. Glutamine utilization by rat neutrophils: Presence of phosphate-dependent glutaminase. Am. J. Physiol. 1997, 273, C1124–C1129. [Google Scholar] [CrossRef]
- Curi, T.C.; de Melo, M.P.; de Azevedo, R.B.; Curi, R. Glutamine utilization by rat neutrophils. Biochem. Soc. Trans. 1997, 25, 249S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pithon-Curi, T.C.; Levada, A.C.; Lopes, L.R.; Doi, S.Q.; Curi, R. Glutamine plays a role in superoxide production and the expression of p47phox, p22phox and gp91phox in rat neutrophils. Clin. Sci. 2002, 103, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Healy, D.A.; Watson, R.W.G.; Newsholme, P. Glucose, but not glutamine, protects against spontaneous and anti-Fas antibody-induced apoptosis in human neutrophils. Clin. Sci. 2002, 103, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef]
- Riffelmacher, T.; Clarke, A.; Richter, F.C.; Stranks, A.; Pandey, S.; Danielli, S.; Hublitz, P.; Yu, Z.; Johnson, E.; Schwerd, T.; et al. Autophagy-Dependent Generation of Free Fatty Acids Is Critical for Normal Neutrophil Differentiation. Immunity 2017, 47, 466–480.e5. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Medina, E.; Hartl, D. Myeloid-Derived Suppressor Cells in Infection: A General Overview. J. Innate Immun. 2018, 10, 407–413. [Google Scholar] [CrossRef]
- Cripps, J.G.; Gorham, J.D. MDSC in autoimmunity. Int. Immunopharmacol. 2011, 11, 789–793. [Google Scholar] [CrossRef]
- Hu, C.; Pang, B.; Lin, G.; Zhen, Y.; Yi, H. Energy metabolism manipulates the fate and function of tumour myeloid-derived suppressor cells. Br. J. Cancer 2020, 122, 23–29. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, N.T.; Kimura, A.; Nakahama, T.; Chinen, I.; Masuda, K.; Nohara, K.; Fujii-Kuriyama, Y.; Kishimoto, T. Aryl hydrocarbon receptor negatively regulates dendritic cell immunogenicity via a kynurenine-dependent mechanism. Proc. Natl. Acad. Sci. USA 2010, 107, 19961–19966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jian, S.-L.; Chen, W.-W.; Su, Y.-C.; Su, Y.-W.; Chuang, T.-H.; Hsu, S.-C.; Huang, L.-R. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Pucino, V.; Bombardieri, M.; Pitzalis, C.; Mauro, C. Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur. J. Immunol. 2017, 47, 14–21. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef]
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e6. [Google Scholar] [CrossRef] [Green Version]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef]



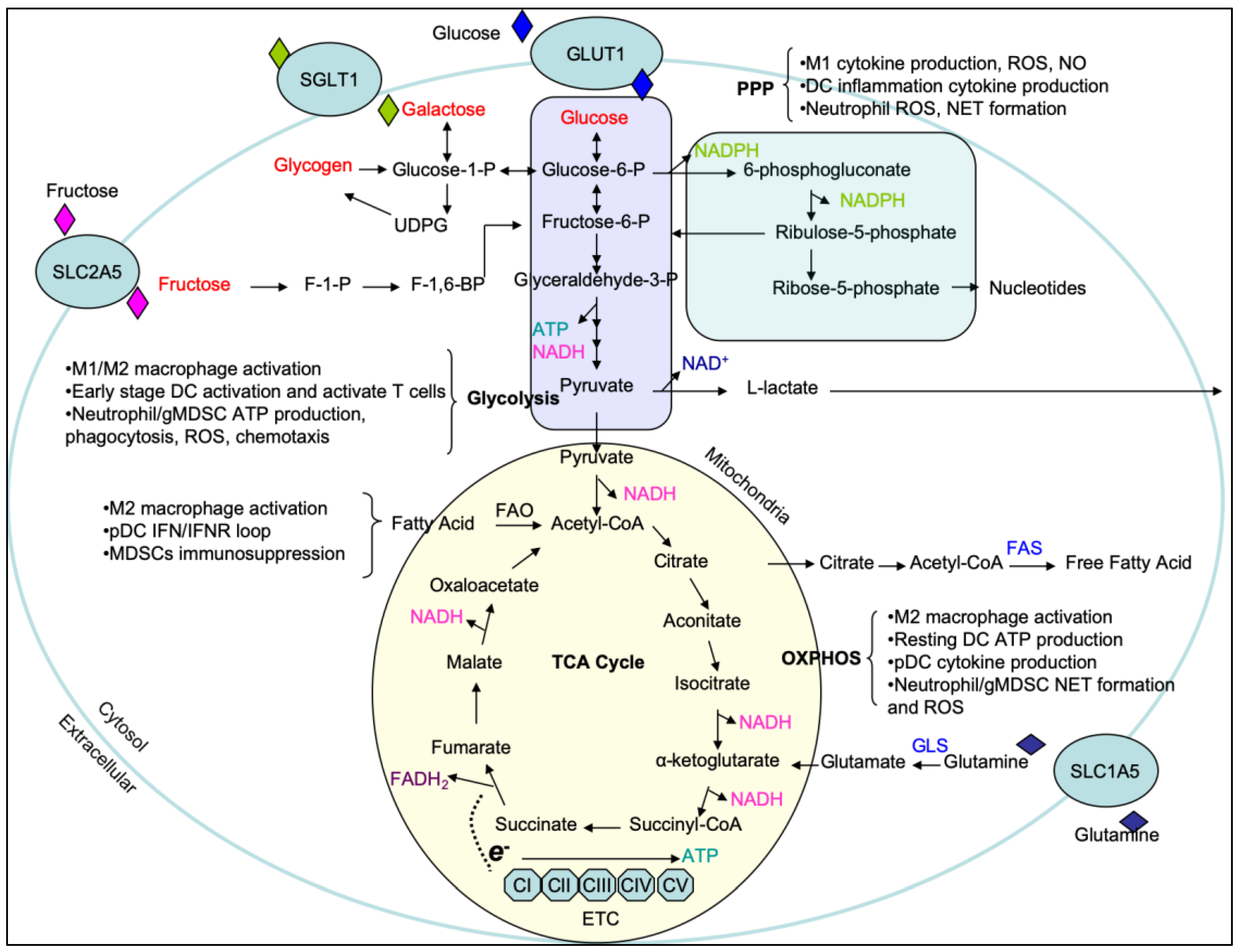{kind=link}
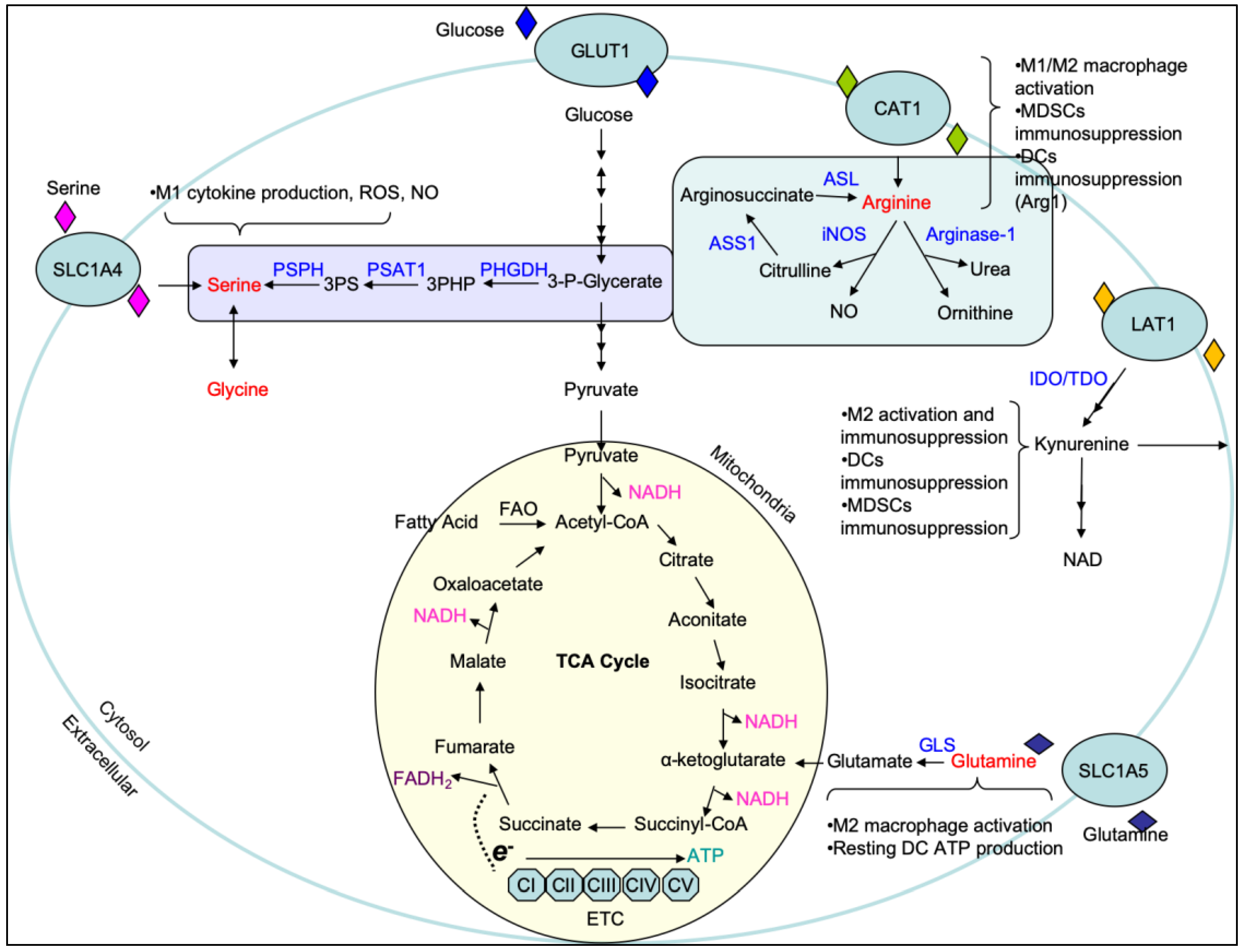{kind=link}
| Cell Type | Inducers | Activation Markers | Cytokine/Chemokine Production | Metabolic Pathways | Cellular Signaling Pathways | Functions |
|---|---|---|---|---|---|---|
| M1 | LPS/IFN-γ | CD80, CD86, MHC-II, CCR7, iNOS | IL-1β, IL-6, IL-12, IL-15, IL-23, TNF-α, CCL3, CCL5, CXCL10 | Glycolysis, PPP, Broken TCA cycle, FAS | NF-kB, PI3K/Akt, mTORC1, HIF-1α, STAT1, IRF5 | Killing intracellular pathogens; Anti-tumor immunity |
| M2 | IL-4/IL-13 | CD206, CD301, PD-L2, RELMα, CD163, Arg1 | IL-10, TGF-β, IL-1Ra, CCL17, CCL22, CCL24 | Glycolysis, OXPHOS/ETC, FAO, Glutaminolysis | PI3K/Akt, AMPK, PGC1β PPAR-γ mTORC1, mTORC2, STAT6, IRF4 | Tissue repair; Anti-helminth immunity; Pro-tumor activity |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, H.; Raines, L.N.; Huang, S.C.-C. Carbohydrate and Amino Acid Metabolism as Hallmarks for Innate Immune Cell Activation and Function. Cells 2020, 9, 562. https://doi.org/10.3390/cells9030562
Zhao H, Raines LN, Huang SC-C. Carbohydrate and Amino Acid Metabolism as Hallmarks for Innate Immune Cell Activation and Function. Cells. 2020; 9(3):562. https://doi.org/10.3390/cells9030562
Chicago/Turabian StyleZhao, Haoxin, Lydia N. Raines, and Stanley Ching-Cheng Huang. 2020. "Carbohydrate and Amino Acid Metabolism as Hallmarks for Innate Immune Cell Activation and Function" Cells 9, no. 3: 562. https://doi.org/10.3390/cells9030562
APA StyleZhao, H., Raines, L. N., & Huang, S. C. -C. (2020). Carbohydrate and Amino Acid Metabolism as Hallmarks for Innate Immune Cell Activation and Function. Cells, 9(3), 562. https://doi.org/10.3390/cells9030562