Insights from In Vivo Studies of Cellular Senescence


{kind=link}
{kind=link}
Abstract
:1. Introduction
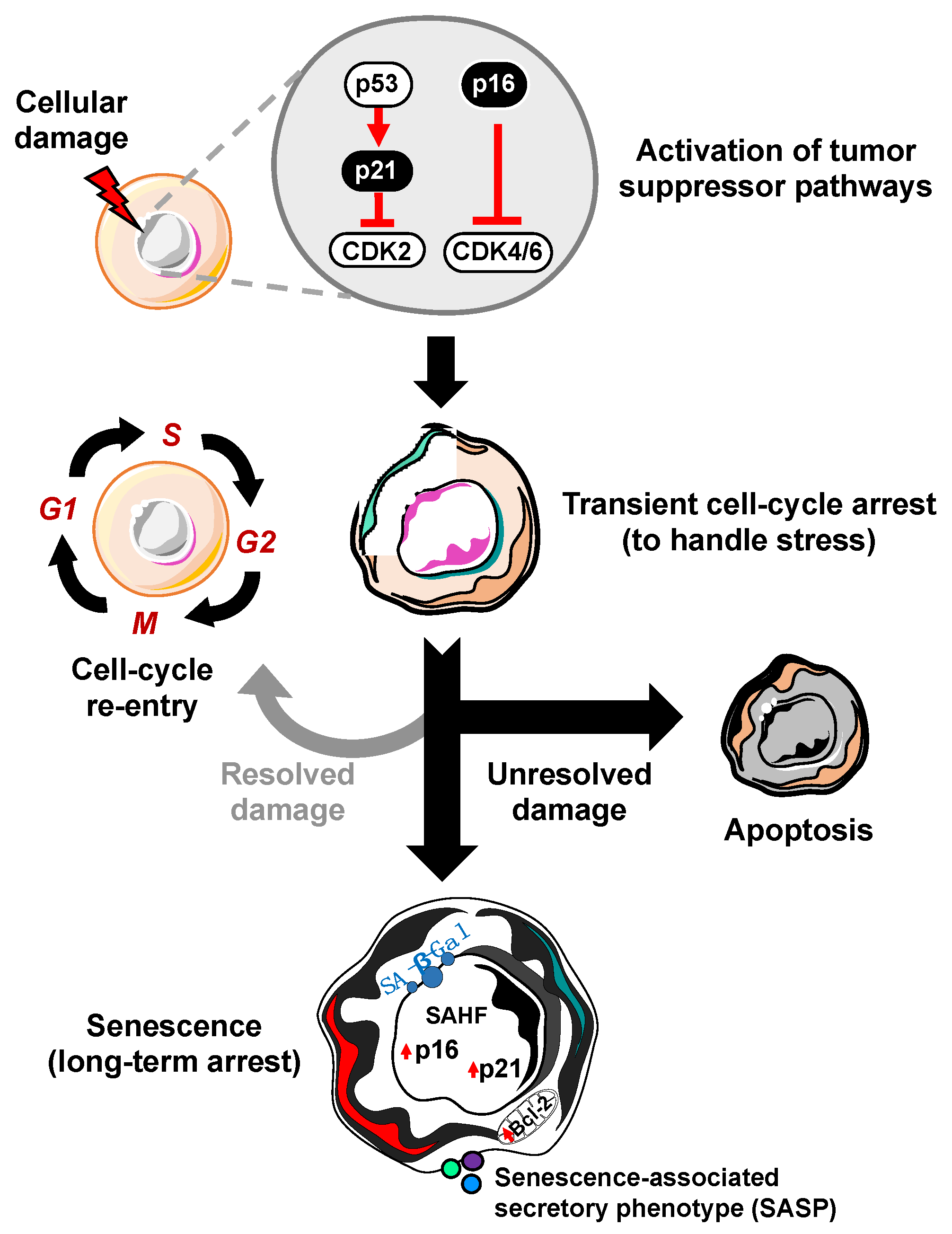
2. Molecular Mechanisms of Cellular Senescence
3. Inducers of Cellular Senescence In Vivo
3.1. Replicative Stress and DNA Damage
3.2. Oncogenic Stress
3.3. Inflammatory Signaling
3.4. Metabolic Stress
4. Cell Type Specificity of Cellular Senescence
5. Senolytics
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science (New York) 2016, 354, 472–477. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science (New York) 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef]
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Beausejour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Yamakoshi, K.; Takahashi, A.; Hirota, F.; Nakayama, R.; Ishimaru, N.; Kubo, Y.; Mann, D.J.; Ohmura, M.; Hirao, A.; Saya, H.; et al. Real-time in vivo imaging of p16Ink4a reveals cross talk with p53. J. Cell Biol. 2009, 186, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Weaver, R.L.; van Deursen, J.M. p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep. 2013, 3, 1164–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Niederlander, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; Hiddinga, H.J.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Prieto, L.I.; Baker, D.J. Cellular Senescence and the Immune System in Cancer. Gerontology 2019, 65, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.Q.; Zhang, Z.Q.; Chen, H.Z.; Liu, D.P. Epigenetic regulation in cell senescence. J. Mol. Med. 2017, 95, 1257–1268. [Google Scholar] [CrossRef]
- Xie, W.; Kagiampakis, I.; Pan, L.; Zhang, Y.W.; Murphy, L.; Tao, Y.; Kong, X.; Kang, B.; Xia, L.; Carvalho, F.L.F.; et al. DNA Methylation Patterns Separate Senescence from Transformation Potential and Indicate Cancer Risk. Cancer Cell 2018, 33, 309–321. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1233. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; McBryan, T.; Jeyapalan, J.C.; Sedivy, J.M.; Adams, P.D. A comparison of oncogene-induced senescence and replicative senescence: Implications for tumor suppression and aging. Age 2014, 36, 9637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Muller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Choudhury, A.R.; Ju, Z.; Djojosubroto, M.W.; Schienke, A.; Lechel, A.; Schaetzlein, S.; Jiang, H.; Stepczynska, A.; Wang, C.; Buer, J.; et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nat. Genet. 2007, 39, 99–105. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braig, M.; Lee, S.; Loddenkemper, C.; Rudolph, C.; Peters, A.H.; Schlegelberger, B.; Stein, H.; Dorken, B.; Jenuwein, T.; Schmitt, C.A. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005, 436, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, C.J.; Keister, B.A.; Stairs, D.B.; Boxer, R.B.; Moody, S.E.; Chodosh, L.A. Dose-dependent oncogene-induced senescence in vivo and its evasion during mammary tumorigenesis. Nat. Cell Biol. 2007, 9, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Neurath, M.F. Resolution of chronic inflammatory disease: Universal and tissue-specific concepts. Nat. Commun. 2018, 9, 3261. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef]
- Bernal, G.M.; Wahlstrom, J.S.; Crawley, C.D.; Cahill, K.E.; Pytel, P.; Liang, H.; Kang, S.; Weichselbaum, R.R.; Yamini, B. Loss of Nfkb1 leads to early onset aging. Aging 2014, 6, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Evans, G.; Tonne, J.M.; Verzosa, G.C.; Stout, M.B.; Mazula, D.L.; Palmer, A.K.; Baker, D.J.; Jensen, M.D.; et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 2016, 65, 1606–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, A.K.; Xu, M.; Zhu, Y.; Pirtskhalava, T.; Weivoda, M.M.; Hachfeld, C.M.; Prata, L.G.; van Dijk, T.H.; Verkade, E.; Casaclang-Verzosa, G.; et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 2019, 18, e12950. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estebanez, B.; de Paz, J.A.; Cuevas, M.J.; Gonzalez-Gallego, J. Endoplasmic Reticulum Unfolded Protein Response, Aging and Exercise: An Update. Front. Physiol. 2018, 9, 1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, P.; Alzahrani, A.M.; Hanieh, H.N.; Kumar, S.A.; Ben Ammar, R.; Rengarajan, T.; Alhoot, M.A. Autophagy and senescence: A new insight in selected human diseases. J. Cell Physiol. 2019, 234, 21485–21492. [Google Scholar] [CrossRef]
- Turi, Z.; Lacey, M.; Mistrik, M.; Moudry, P. Impaired ribosome biogenesis: Mechanisms and relevance to cancer and aging. Aging 2019, 11, 2512–2540. [Google Scholar] [CrossRef]
- Liu, J.Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Burd, C.E.; Sorrentino, J.A.; Clark, K.S.; Darr, D.B.; Krishnamurthy, J.; Deal, A.M.; Bardeesy, N.; Castrillon, D.H.; Beach, D.H.; Sharpless, N.E. Monitoring tumorigenesis and senescence in vivo with a p16(INK4a)-luciferase model. Cell 2013, 152, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.I.; et al. p16(Ink4a) and senescence-associated beta-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging 2017, 9, 1867–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Zhang, K.; Chen, H.; Zhao, X.; Wang, J.; Li, L.; Cong, Y.; Ju, Z.; Xu, D.; Williams, B.R.; et al. Telomerase Deficiency Causes Alveolar Stem Cell Senescence-associated Low-grade Inflammation in Lungs. J. Biol. Chem. 2015, 290, 30813–30829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.L.; Kracht, M. Cyclin-Dependent Kinases as Coregulators of Inflammatory Gene Expression. Trends Pharmacol. Sci. 2016, 37, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.I.; Nishio, N.; Hasegawa, T. Immunological aspects of age-related diseases. World J. Biol. Chem. 2017, 8, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Birch, J.; Passos, J.F. Targeting the SASP to combat ageing: Mitochondria as possible intracellular allies? Bioessays 2017, 39. [Google Scholar] [CrossRef] [Green Version]
- Korolchuk, V.I.; Miwa, S.; Carroll, B.; von Zglinicki, T. Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 2017, 21, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Momcilovic, M.; Jones, A.; Bailey, S.T.; Waldmann, C.M.; Li, R.; Lee, J.T.; Abdelhady, G.; Gomez, A.; Holloway, T.; Schmid, E.; et al. In vivo imaging of mitochondrial membrane potential in non-small-cell lung cancer. Nature 2019, 575, 380–384. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz-Espin, D.; Rovira, M.; Galiana, I.; Gimenez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Munoz-Martin, M.; Ucero, A.C.; et al. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto, L.I.; Graves, S.I.; Baker, D.J. Insights from In Vivo Studies of Cellular Senescence. Cells 2020, 9, 954. https://doi.org/10.3390/cells9040954
Prieto LI, Graves SI, Baker DJ. Insights from In Vivo Studies of Cellular Senescence. Cells. 2020; 9(4):954. https://doi.org/10.3390/cells9040954
Chicago/Turabian StylePrieto, Luis I., Sara I. Graves, and Darren J. Baker. 2020. "Insights from In Vivo Studies of Cellular Senescence" Cells 9, no. 4: 954. https://doi.org/10.3390/cells9040954
APA StylePrieto, L. I., Graves, S. I., & Baker, D. J. (2020). Insights from In Vivo Studies of Cellular Senescence. Cells, 9(4), 954. https://doi.org/10.3390/cells9040954