FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARγ Specific Regulation: A Transcriptome Analysis

 , , , ,
, , , ,  , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement and Obtained Samples, Isolation, and Differentiation of hASCs
2.2. Flow Cytometry
2.3. RNA and DNA Isolation, Genotyping
2.4. RNA-Sequencing
2.5. Antibodies and Immunoblotting
2.6. Immunofluorescence Staining, Quantification of Browning
2.7. Determination of Cellular Oxygen Consumption (OC) and Extracellular Acidification Rate (ECAR)
2.8. Statistical Analysis
3. Results
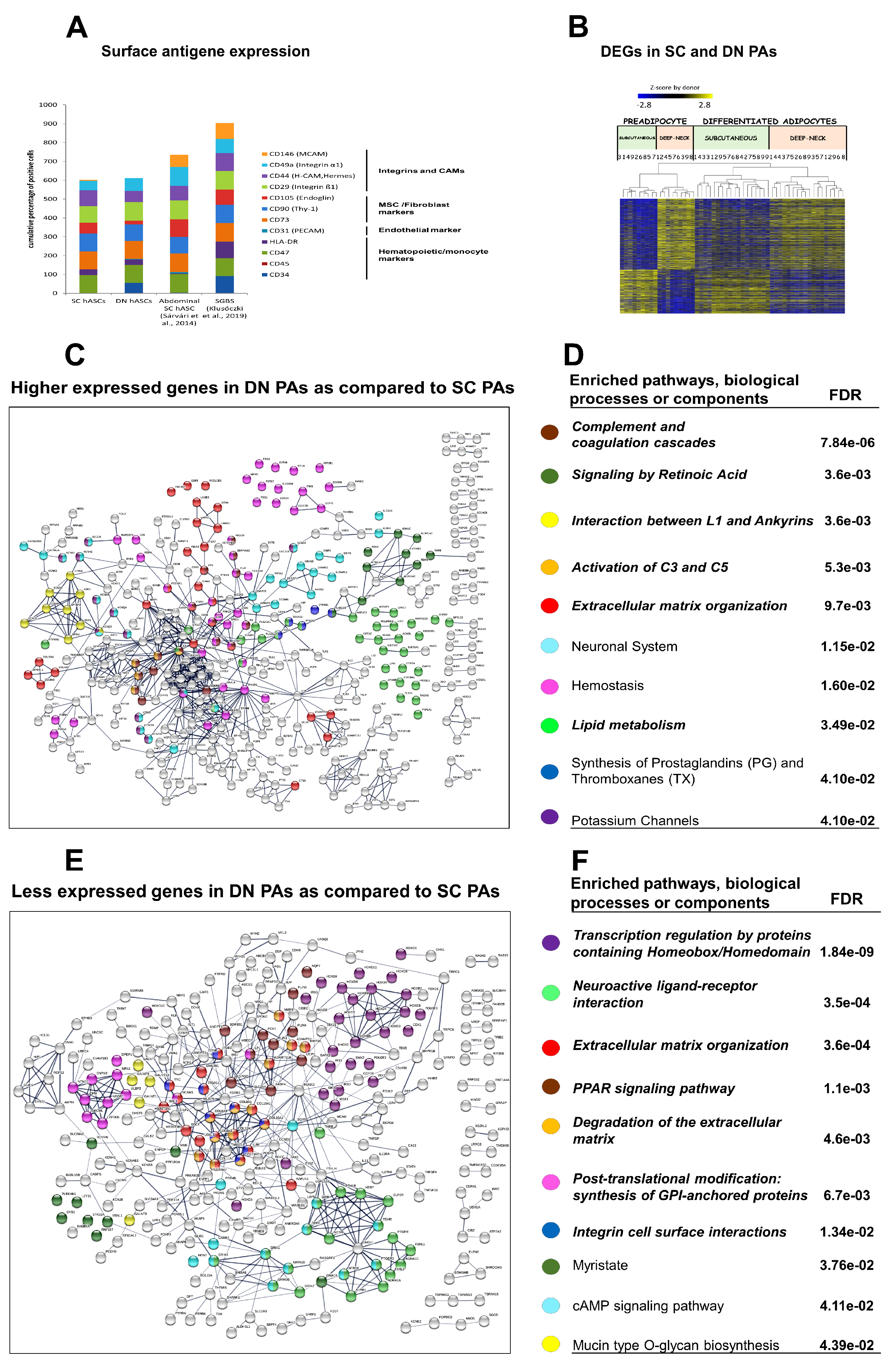
3.1. DN and SC Progenitors Have Similar Surface Markers and Adipocyte Differentiation Potential But Differ in the Gene Expression Profile
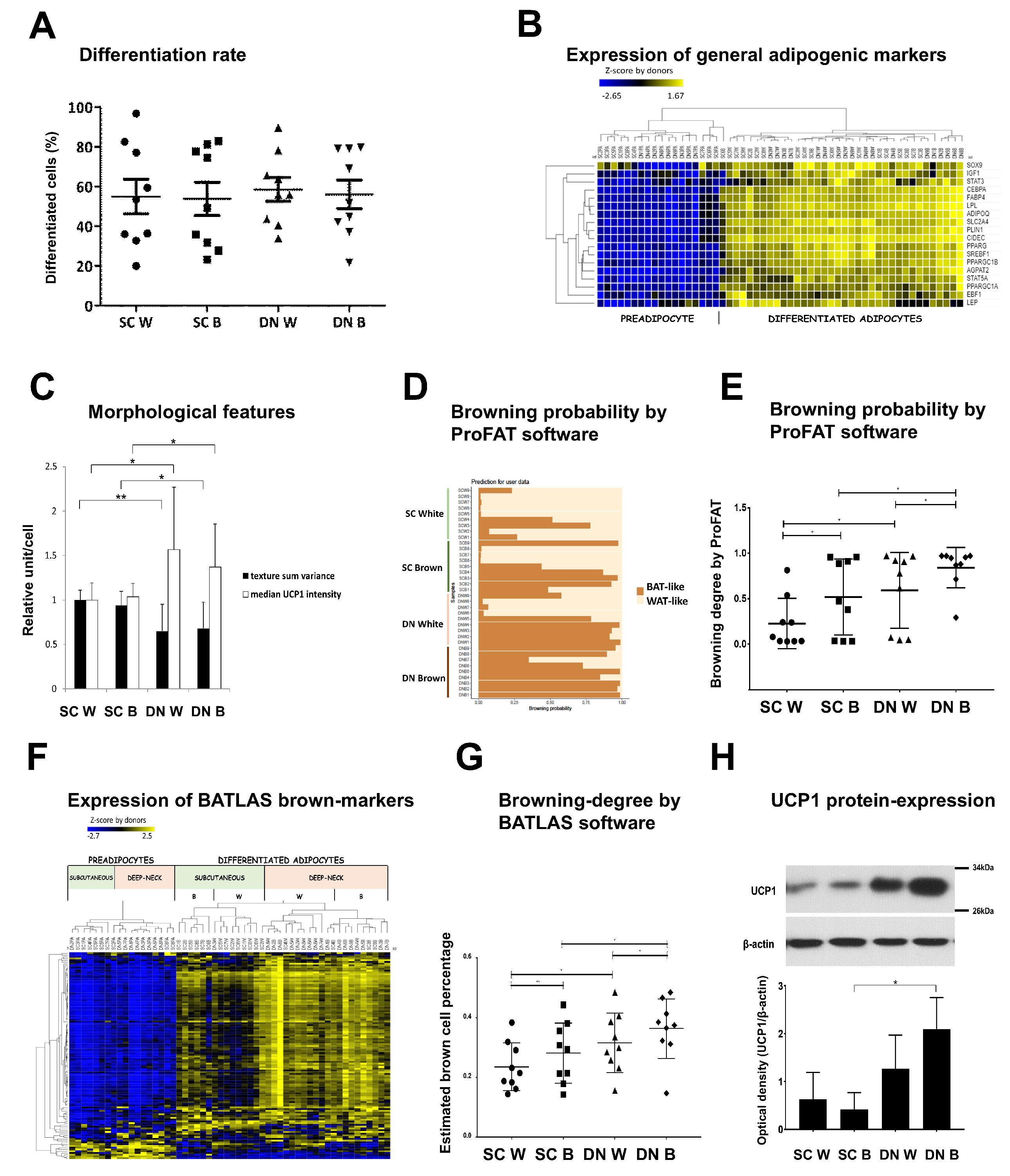
3.2. Differentiated Adipocytes from DN Progenitors Display Higher Browning-Related Gene-Expression Features
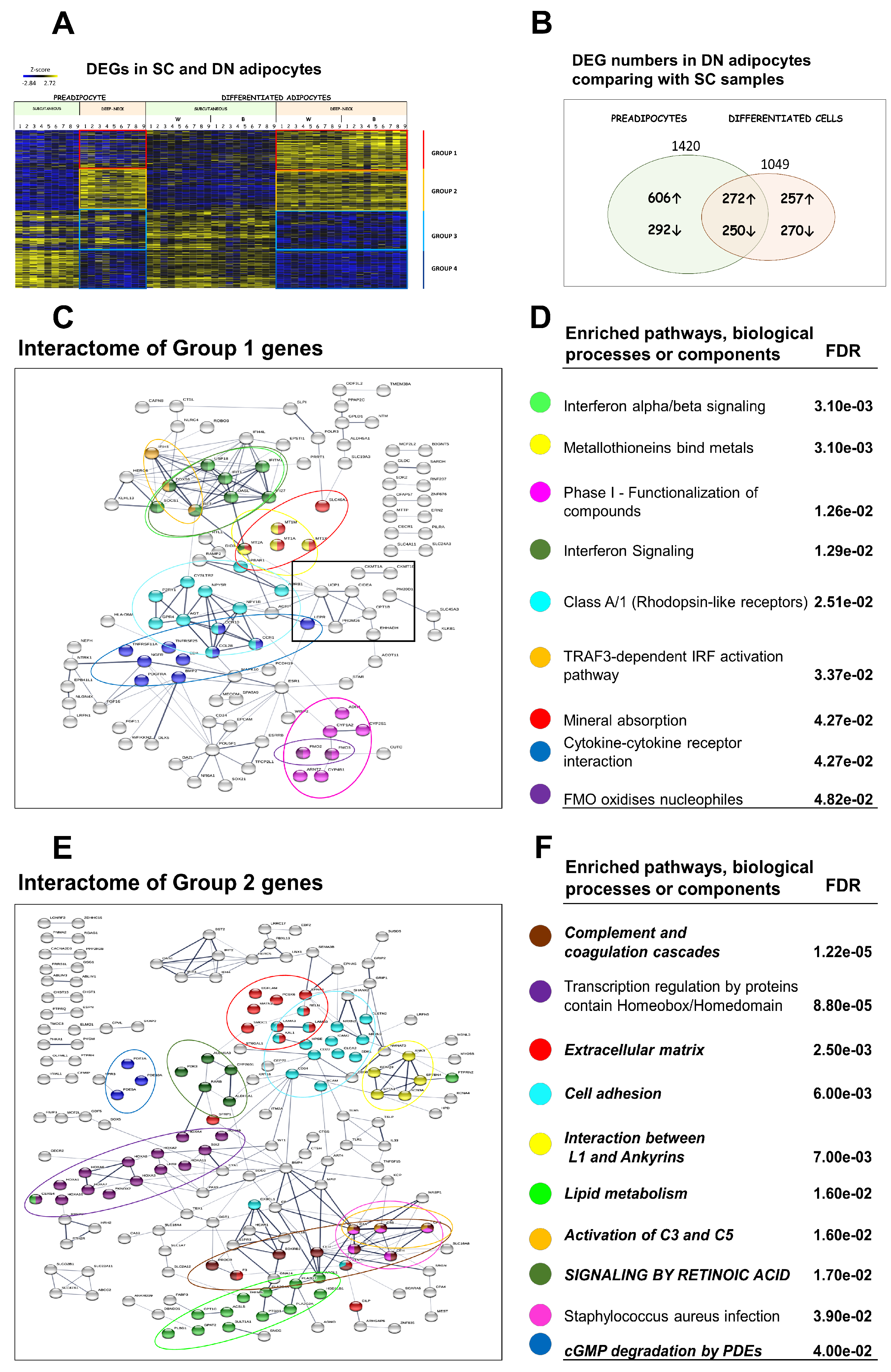
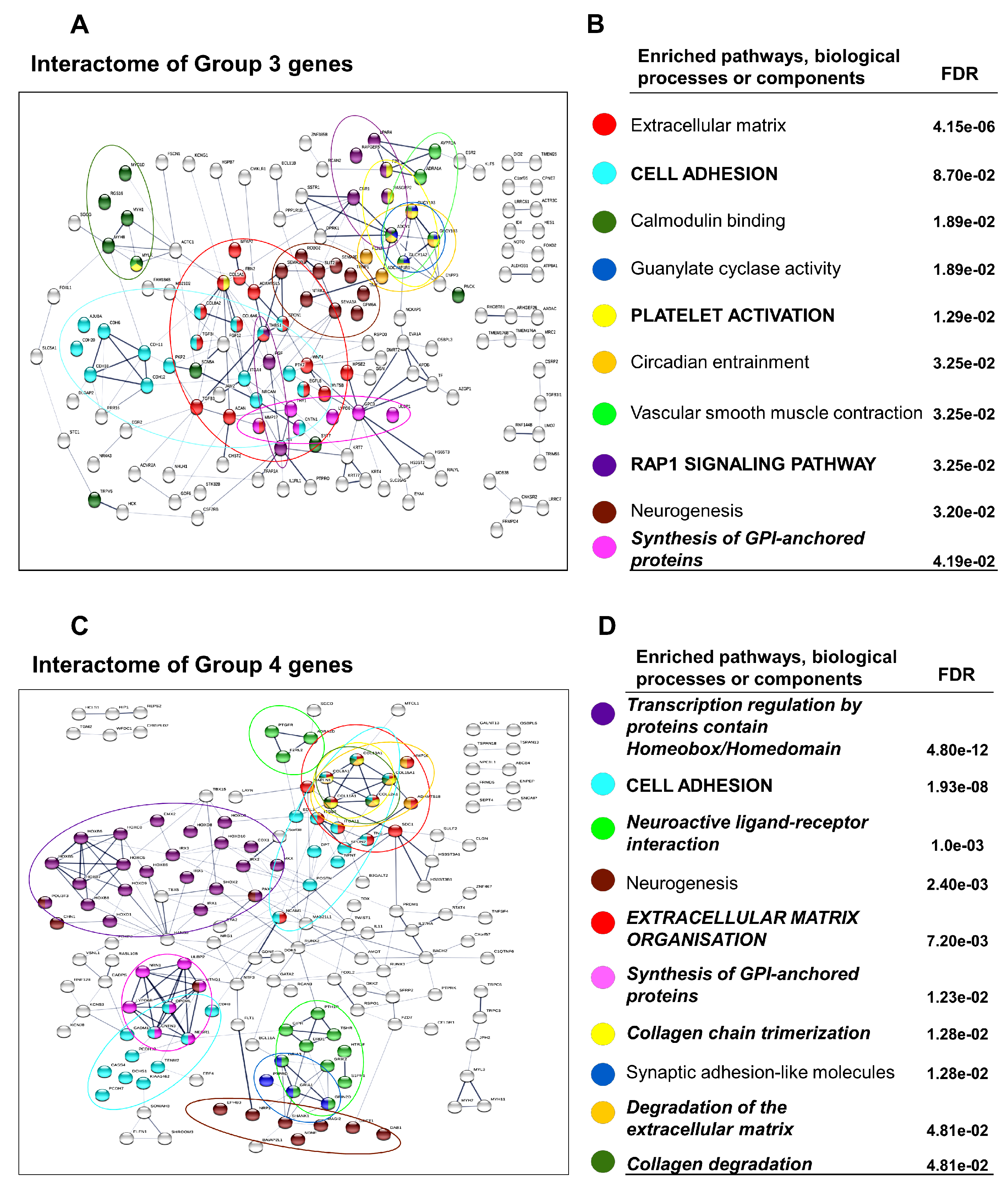
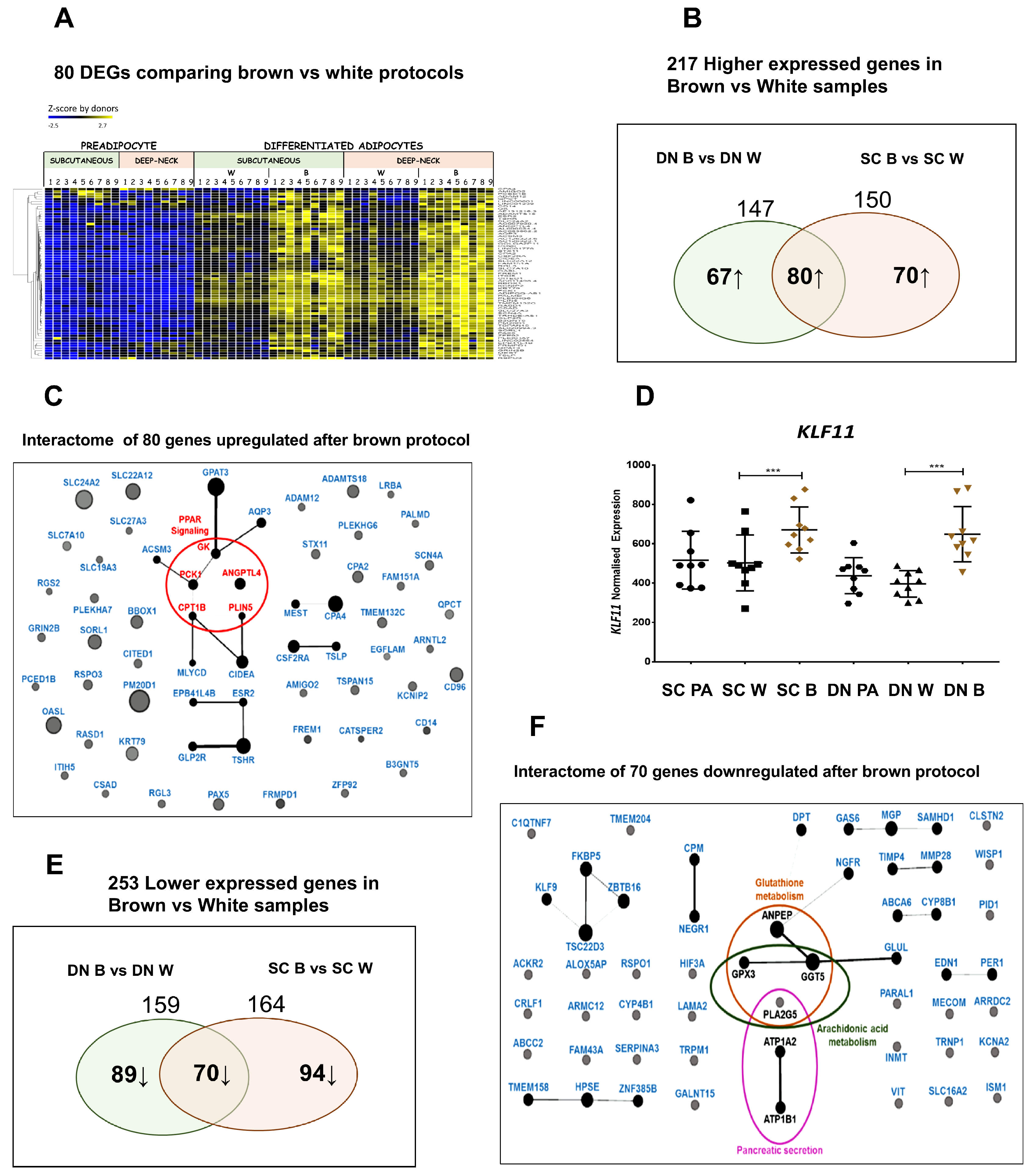
3.3. Differential Gene Expression Patterns Reveal Network Pathways Associated with Higher Browning Potential of DN Adipocytes
3.4. Downregulated Pathways in DN Adipocytes
3.5. Shared PPARγ Mediated Gene Expression Patterns in DN and SC Adipocytes
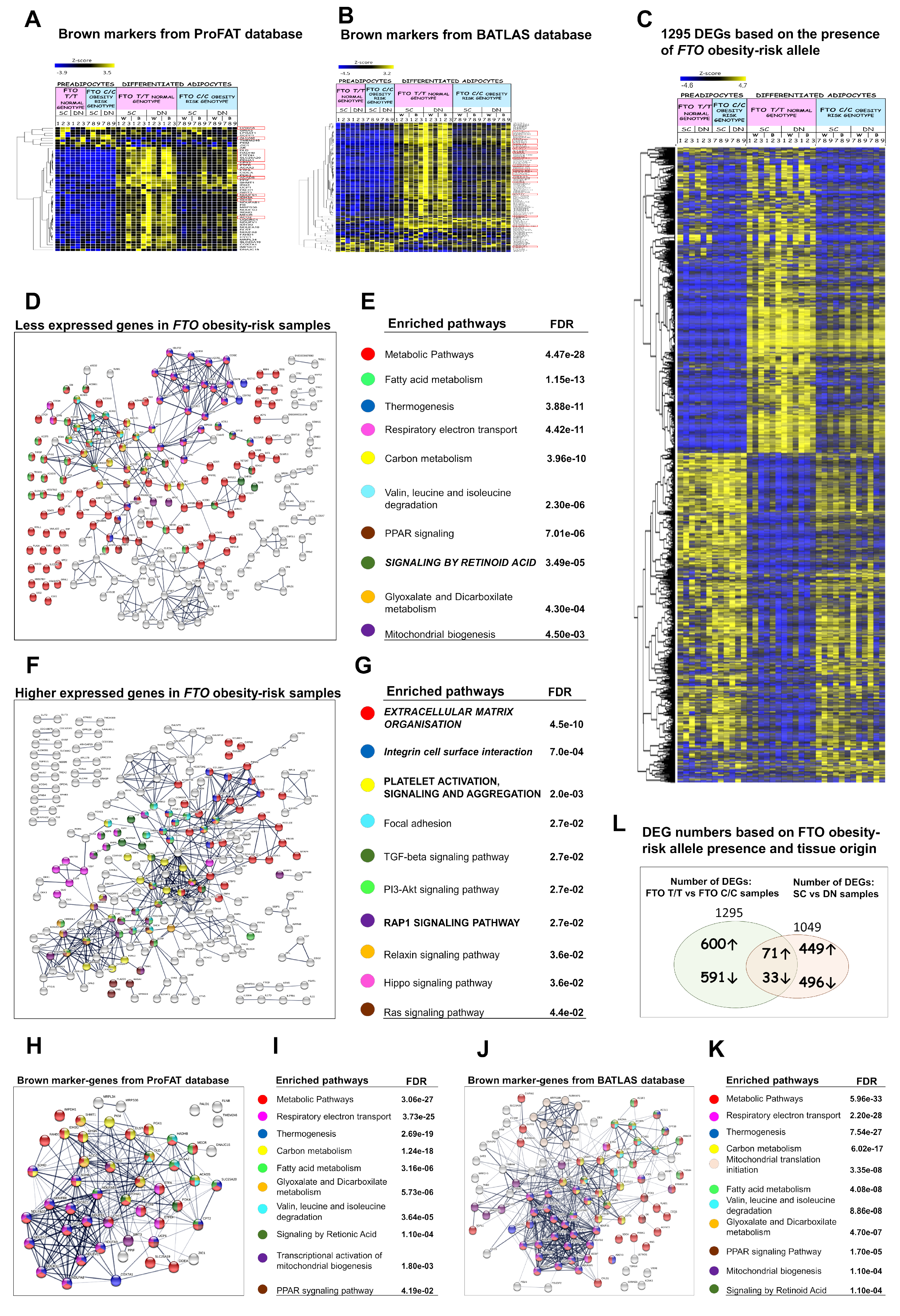
3.6. Presence of the FTO Obesity-Risk Alleles Significantly Influences Browning Gene Expression Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cannon, B.; Nedergaard, J. Brown Adipose Tissue: Function and Physiological Significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of Fatty-Acid-Dependent UCP1 Uncoupling in Brown Fat Mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazak, L.; Chouchani, E.T.; Lu, G.Z.; Jedrychowski, M.P.; Bare, C.J.; Mina, A.I.; Kumari, M.; Zhang, S.; Vuckovic, I.; Laznik-Bogoslavski, D.; et al. Genetic Depletion of Adipocyte Creatine Metabolism Inhibits Diet-Induced Thermogenesis and Drives Obesity. Cell Metab. 2017, 26, 660–671.e3. [Google Scholar] [CrossRef] [Green Version]
- Kristof, E.; Doan-Xuan, Q.-M.; Sárvári, A.K.; Klusóczki, Á.; Fischer-Posovszky, P.; Wabitsch, M.; Bacso, Z.; Bai, P.; Balajthy, Z.; Fésüs, L. Clozapine modifies the differentiation program of human adipocytes inducing browning. Transl. Psychiatry 2016, 6, e963. [Google Scholar] [CrossRef]
- Klusóczki, Á.; Veréb, Z.; Vámos, A.; Fischer-Posovszky, P.; Wabitsch, M.; Bacso, Z.; Fésüs, L.; Kristof, E. Differentiating SGBS adipocytes respond to PPARγ stimulation, irisin and BMP7 by functional browning and beige characteristics. Sci. Rep. 2019, 9, 5823. [Google Scholar]
- Ikeda, K.; Kang, Q.; Yoneshiro, T.; Camporez, J.P.; Maki, H.; Homma, M.; Shinoda, K.; Chen, Y.; Lu, X.; Maretich, P.; et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med. 2017, 23, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- Long, J.Z.; Svensson, K.J.; Bateman, L.A.; Lin, H.; Kamenecka, T.; Lokurkar, I.A.; Lou, J.; Rao, R.R.; Chang, M.R.; Jedrychowski, M.P.; et al. The Secreted Enzyme PM20D1 Regulates Lipidated Amino Acid Uncouplers of Mitochondria. Cell 2016, 166, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef] [Green Version]
- Whittle, A.; López, M.; Vidal-Puig, A. Using brown adipose tissue to treat obesity – the central issue. Trends Mol. Med. 2011, 17, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scimè, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, N.; Walden, T.B.; Shabalina, I.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Chronic Peroxisome Proliferator-activated Receptor γ (PPARγ) Activation of Epididymally Derived White Adipocyte Cultures Reveals a Population of Thermogenically Competent, UCP1-containing Adipocytes Molecularly Distinct from Classic Brown Adipocytes. J. Biol. Chem. 2009, 285, 7153–7164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leitner, B.P.; Huang, S.; Brychta, R.J.; Duckworth, C.J.; Baskin, A.S.; McGehee, S.; Tal, I.; Dieckmann, W.; Gupta, G.; Kolodny, G.M.; et al. Mapping of human brown adipose tissue in lean and obese young men. Proc. Natl. Acad. Sci. USA 2017, 114, 8649–8654. [Google Scholar] [CrossRef] [Green Version]
- Svensson, P.-A.; Jernås, M.; Sjöholm, K.; Hoffmann, J.M.; Nilsson, B.E.; Hansson, M.; Carlsson, L.M. Gene expression in human brown adipose tissue. Int. J. Mol. Med. 2011, 27, 227–232. [Google Scholar] [CrossRef]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Wu, J.; Boström, P.; Sparks, L.; Ye, L.; Choi, J.H.; Giang, A.-H.; Khandekar, M.; A Virtanen, K.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Jespersen, N.; Larsen, T.J.; Peijs, L.; Daugaard, S.; Homøe, P.; Loft, A.; De Jong, J.; Mathur, N.; Cannon, B.; Nedergaard, J.; et al. A Classical Brown Adipose Tissue mRNA Signature Partly Overlaps with Brite in the Supraclavicular Region of Adult Humans. Cell Metab. 2013, 17, 798–805. [Google Scholar] [CrossRef] [Green Version]
- Claussnitzer, M.; Dankel, S.N.; Kim, K.-H.; Quon, G.; Meuleman, W.; Haugen, C.; Glunk, V.; Sousa, I.S.; Beaudry, J.L.; Puviindran, V.; et al. FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N. Engl. J. Med. 2015, 373, 895–907. [Google Scholar] [CrossRef] [Green Version]
- Min, S.Y.; Desai, A.; Yang, Z.; Sharma, A.; DeSouza, T.; Genga, R.M.J.; Kucukural, A.; Lifshitz, L.M.; Nielsen, S.; Scheele, C.; et al. Diverse repertoire of human adipocyte subtypes develops from transcriptionally distinct mesenchymal progenitor cells. Proc. Natl. Acad. Sci. USA 2019, 116, 17970–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdikari, A.; Leparc, G.G.; Balaz, M.; Pires, N.D.; Lidell, M.E.; Sun, W.; Fernandez-Albert, F.; Müller, S.; Akchiche, N.; Dong, H.; et al. BATLAS: Deconvoluting Brown Adipose Tissue. Cell Rep. 2018, 25, 784–797.e4. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Jiang, L.; Keipert, S.; Zhang, S.; Hauser, A.; Graf, E.; Strom, T.; Tschöp, M.; Jastroch, M.; Perocchi, F. Prediction of Adipose Browning Capacity by Systematic Integration of Transcriptional Profiles. Cell Rep. 2018, 23, 3112–3125. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Karlsson, T.; Ek, W.E.; Johansson, Å. Genome-wide association study of body fat distribution identifies adiposity loci and sex-specific genetic effects. Nat. Commun. 2019, 10, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallberg, M.; Morganstein, D.L.; Kiskinis, E.; Shah, K.; Kralli, A.; Dilworth, S.M.; White, R.; Parker, M.G.; Christian, M. A Functional Interaction between RIP140 and PGC-1α Regulates the Expression of the Lipid Droplet Protein CIDEA. Mol. Cell. Biol. 2008, 28, 6785–6795. [Google Scholar] [CrossRef] [Green Version]
- Sharp, L.Z.; Shinoda, K.; Ohno, H.; Scheel, D.W.; Tomoda, E.; Ruiz, L.; Hu, H.; Wang, L.; Pavlova, Z.; Gilsanz, V.; et al. Human BAT Possesses Molecular Signatures That Resemble Beige/Brite Cells. PLoS ONE 2012, 7, e49452. [Google Scholar] [CrossRef] [PubMed]
- Kristóf, E.; Klusóczki, Á.; Veress, R.; Shaw, A.; Combi, Z.S.; Varga, K.; Győry, F.; Balajthy, Z.; Bai, P.; Bacso, Z.; et al. Interleukin-6 released from differentiating human beige adipocytes improves browning. Exp. Cell Res. 2019, 377, 47–55. [Google Scholar] [CrossRef]
- Kristof, E.; Doan-Xuan, Q.-M.; Bai, P.; Bacso, Z.; Fésüs, L. Laser-scanning cytometry can quantify human adipocyte browning and proves effectiveness of irisin. Sci. Rep. 2015, 5, 12540. [Google Scholar] [CrossRef]
- Fischer-Posovszky, P.; Newell, F.S.; Wabitsch, M.; Tornqvist, H.E. Human SGBS cells - a unique tool for studies of human fat cell biology. Obes. Facts 2008, 1, 184–189. [Google Scholar] [CrossRef]
- Elabd, C.; Chiellini, C.; Carmona, M.; Galitzky, J.; Cochet, O.; Petersen, R.; Penicaud, L.; Kristiansen, K.; Bouloumié, A.; Casteilla, L.; et al. Human Multipotent Adipose-Derived Stem Cells Differentiate into Functional Brown Adipocytes. Stem Cells 2009, 27, 2753–2760. [Google Scholar] [CrossRef]
- Sarvari, A.K.; Veréb, Z.J.; Uray, I.P.; Fésüs, L.; Balajthy, Z. Atypical antipsychotics induce both proinflammatory and adipogenic gene expression in human adipocytes in vitro. Biochem. Biophys. Res. Commun. 2014, 450, 1383–1389. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doan-Xuan, Q.M.; Sarvari, A.K.; Wabitsch, M.; Fischer-Posovszky, P.; Balajthy, Z.; Fésüs, L.; Bacso, Z. High content analysis of differentiation and cell death in human adipocytes. Cytom. Part A 2013, 83, 933–943. [Google Scholar]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Tews, D.; Schwar, V.; Scheithauer, M.; Weber, T.; Fromme, T.; Klingenspor, M.; Barth, T.F.E.; Moller, P.; Holzmann, K.; Debatin, K.-M.; et al. Comparative gene array analysis of progenitor cells from human paired deep neck and subcutaneous adipose tissue. Mol. Cell. Endocrinol. 2014, 395, 41–50. [Google Scholar] [CrossRef]
- Lowe, C.E.; O’Rahilly, S.; Rochford, J.J. Adipogenesis at a glance. J. Cell Sci. 2011, 124, 2681–2686. [Google Scholar] [CrossRef] [Green Version]
- Blázquez-Medela, A.; Jumabay, M.; Boström, K.I. Beyond the bone: Bone morphogenetic protein signaling in adipose tissue. Obes. Rev. 2019, 20, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.-W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Loft, A.; Forss, I.; Siersbæk, M.; Schmidt, S.F.; Larsen, A.-S.; Madsen, J.G.S.; Pisani, D.F.; Nielsen, R.; Aagaard, M.M.; Mathison, A.; et al. Browning of human adipocytes requires KLF11 and reprogramming of PPARγ superenhancers. Genes Dev. 2014, 29, 7–22. [Google Scholar] [CrossRef] [Green Version]
- Benson, K.K.; Hu, W.; Weller, A.H.; Bennett, A.H.; Chen, E.R.; Khetarpal, S.A.; Yoshino, S.; Bone, W.P.; Wang, L.; Rabinowitz, J.D.; et al. Natural human genetic variation determines basal and inducible expression of PM20D1, an obesity-associated gene. Proc. Natl. Acad. Sci. USA 2019, 116, 23232–23242. [Google Scholar] [CrossRef] [PubMed]
- Simcox, J.; Geoghegan, G.; Maschek, J.A.; Bensard, C.L.; Pasquali, M.; Miao, R.; Lee, S.; Jiang, L.; Huck, I.; Kershaw, E.E.; et al. Global Analysis of Plasma Lipids Identifies Liver-Derived Acylcarnitines as a Fuel Source for Brown Fat Thermogenesis. Cell Metab. 2017, 26, 509–522.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo, S.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Lu, P.; Shi, J.; Liu, W.; Yang, M.; Zhao, S.; Chen, N.; Chen, M.; Sun, Y.; Gao, A.; et al. IRX3 Promotes the Browning of White Adipocytes and Its Rare Variants are Associated with Human Obesity Risk. EBioMedicine 2017, 24, 64–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Shinoda, K.; Luijten, I.H.N.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.-H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [Green Version]
- Xue, R.; Lynes, M.D.; Dreyfuss, J.M.; Shamsi, F.; Schulz, T.; Zhang, H.; Huang, T.L.; Townsend, K.L.; Li, Y.; Takahashi, H.; et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 2015, 21, 760–768. [Google Scholar] [CrossRef]
- Divoux, A.; Sandor, K.; Bojcsuk, D.; Talukder, A.; Li, X.; Balint, B.L.; Osborne, T.F.; Smith, S.R. Differential open chromatin profile and transcriptomic signature define depot-specific human subcutaneous preadipocytes: Primary outcomes. Clin. Epigenetics 2018, 10, 148. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Fu, X.; Liang, X.; DeAvila, J.M.; Wang, Z.; Zhao, L.; Tian, Q.; Zhao, J.; Gomez, N.A.; Trombetta, S.C.; et al. Retinoic acid induces white adipose tissue browning by increasing adipose vascularity and inducing beige adipogenesis of PDGFRα+ adipose progenitors. Cell Discov. 2017, 3, 17036. [Google Scholar] [CrossRef] [PubMed]
- Mercader, J.; Ribot, J.; Murano, I.; Felipe, F.; Cinti, S.; Bonet, M.L.; Palou, A. Remodeling of White Adipose Tissue after Retinoic Acid Administration in Mice. Endocrinology 2006, 147, 5325–5332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kissig, M.; Rajakumari, S.; Huang, L.; Lim, H.-W.; Won, K.-J.; Seale, P. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 14466–14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, R.R.; Shapira, S.N.; Lim, H.-W.; Ishibashi, J.; Harms, M.; Won, K.-J.; Seale, P. EBF2 promotes the recruitment of beige adipocytes in white adipose tissue. Mol. Metab. 2015, 5, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef] [Green Version]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional Control of Brown Fat Determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tvrdik, P.; Asadi, A.; Kozak, L.P.; Nedergaard, J.; Cannon, B.; Jacobsson, A. Cig30, a Mouse Member of a Novel Membrane Protein Gene Family, Is Involved in the Recruitment of Brown Adipose Tissue. J. Biol. Chem. 1997, 272, 31738–31746. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Park, A.; Kim, W.K.; Bae, K.-H. Distinction of white, beige and brown adipocytes derived from mesenchymal stem cells. World J. Stem Cells 2014, 6, 33–42. [Google Scholar] [CrossRef]
- Villarroya, J.; Cereijo, R.; Gavaldà-Navarro, A.; Peyrou, M.; Giralt, M.; Villarroya, F. New insights into the secretory functions of brown adipose tissue. J. Endocrinol. 2019, 243, R19–R27. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, F.E.; Carrión, F.; Villanueva, S.; Khoury, M. Mesenchymal Stem Cell treatment for autoimmune diseases: A critical review. Biol. Res. 2012, 45, 269–277. [Google Scholar] [CrossRef] [Green Version]
- Tatullo, M.; Codispoti, B.; Pacifici, A.; Palmieri, F.; Marrelli, M.; Pacifici, L.; Paduano, F. Potential Use of Human Periapical Cyst-Mesenchymal Stem Cells (hPCy-MSCs) as a Novel Stem Cell Source for Regenerative Medicine Applications. Front. Cell Dev. Biol. 2017, 5, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsen-Kanson, T.; Hafner, A.-L.; Wdziekonski, B.; Takashima, Y.; Villageois, P.; Carrière, A.; Svensson, M.; Bagnis, C.; Chignon-Sicard, B.; Svensson, P.-A.; et al. Differentiation of Human Induced Pluripotent Stem Cells into Brown and White Adipocytes: Role of Pax3. Stem Cells 2014, 32, 1459–1467. [Google Scholar] [CrossRef] [PubMed]
- Bjune, J.-I.; Haugen, C.; Gudbrandsen, O.; Nordbø, O.P.; Nielsen, H.J.; Våge, V.; Njølstad, P.R.; Sagen, J.V.; Dankel, S.N.; Mellgren, G. IRX5 regulates adipocyte amyloid precursor protein and mitochondrial respiration in obesity. Int. J. Obes. 2018, 43, 2151–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smemo, S.; Tena, J.J.; Kim, K.-H.; Gamazon, E.R.; Sakabe, N.J.; Gómez-Marín, C.; Aneas, I.; Credidio, F.L.; Sobreira, D.R.; Wasserman, N.F.; et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 2014, 507, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Hartley, B.J.; Scott, D.J.; Callander, G.E.; Wilkinson, T.N.; Ganella, D.E.; Kong, C.K.; Layfield, S.; Ferraro, T.; Petrie, E.; Bathgate, R.A.D. Resolving the Unconventional Mechanisms Underlying RXFP1 and RXFP2 Receptor Function. Ann. N. Y. Acad. Sci. 2009, 1160, 67–73. [Google Scholar] [CrossRef]
- Torii, S.; Kubota, C.; Saito, N.; Kawano, A.; Hou, N.; Kobayashi, M.; Torii, R.; Hosaka, M.; Kitamura, T.; Takeuchi, T.; et al. The pseudophosphatase phogrin enables glucose-stimulated insulin signaling in pancreatic β cells. J. Biol. Chem. 2018, 293, 5920–5933. [Google Scholar] [CrossRef] [Green Version]
- Gomi, H.; Kubota-Murata, C.; Yasui, T.; Tsukise, A.; Torii, S. Immunohistochemical Analysis of IA-2 Family of Protein Tyrosine Phosphatases in Rat Gastrointestinal Endocrine Cells. J. Histochem. Cytochem. 2012, 61, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Jain, D.; Liu, Q.; Bartosinska, B.; Wang, J.; Schumann, D.M.; Kauschke, S.G.; Eickelmann, P.; Piemonti, L.; Gray, N.S.; et al. Pharmacological inhibition of Eph receptors enhances glucose-stimulated insulin secretion from mouse and human pancreatic islets. Diabetologia 2013, 56, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Barrett, N.A.; Kanaoka, Y.; Yoshimoto, E.; Garofalo, D.; Cirka, H.; Feng, C.; Boyce, J.A. Type 2 Cysteinyl Leukotriene Receptors Drive IL-33–Dependent Type 2 Immunopathology and Aspirin Sensitivity. J. Immunol. 2017, 200, 915–927. [Google Scholar] [CrossRef]
- Qiu, Y.; Nguyen, K.; Odegaard, J.I.; Cui, X.; Tian, X.Y.; Locksley, R.M.; Palmiter, R.D.; Chawla, A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014, 157, 1292–1308. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; Van Der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C.; et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623–630. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Gene | BC Score | Bridges | FC | Gene Description |
|---|---|---|---|---|---|
| 1 | ESR1 | 1445 | 3 | 2.0 | estrogen receptor 1 |
| UCP1 | 1323 | 1 | 6.1 | uncoupling protein 1 (mitochondrial, proton carrier) | |
| MT2A | 1061 | 2 | 2.4 | metallothionein 2A | |
| LEPR | 988 | 1 | 1.9 | leptin receptor | |
| IFIH1 | 957 | 0 | 2.0 | interferon induced with helicase C domain 1 | |
| IRF7 | 880 | 2 | 2.0 | interferon regulatory factor 7 | |
| NLRC4 | 864 | 1 | 1.9 | NLR family, CARD domain containing 4 | |
| CTSL | 813 | 1 | 1.9 | cathepsin L | |
| AGT | 745 | 2 | 2.4 | angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | |
| MAPK10 | 720 | 2 | 1.8 | mitogen-activated protein kinase 10 | |
| 2 | BMP4 | 2776 | 8 | 3.1 | bone morphogenetic protein 4 |
| CD34 | 2048 | 3 | 3.6 | stem cell adhesion | |
| EFNA5 | 1519 | 1 | 2.2 | ephrin-A5 | |
| RARB | 1362 | 0 | 3.9 | retinoic acid receptor, beta | |
| ALDH1A3 | 1194 | 1 | 4.4 | aldehyde dehydrogenase 1 family, member A3 | |
| SMOC1 | 1138 | 1 | 3.2 | SPARC related modular calcium binding 1 | |
| TBX1 | 1125 | 3 | 2.5 | T-box 1 | |
| RELN | 1087 | 0 | 3.1 | reelin | |
| SEMA3B | 1050 | 1 | 2.2 | immunoglobulin domain (semaphorin) short basic domain, secreted, 3B | |
| SPTA1 | 1046 | 1 | 2.1 | spectrin, alpha, erythrocytic 1 (elliptocytosis 2) |
| Group | Gene | BC Score | Bridges | FC | Gene Description |
|---|---|---|---|---|---|
| 3 | COL1A1 | 2057 | 4 | 2.1 | collagen, type I, alpha 1 |
| GPC3 | 1113 | 1 | 3.0 | glypican 3 | |
| KRT7 | 914 | 2 | 2.9 | keratin 7 | |
| CNR1 | 864 | 1 | 5.8 | cannabinoid receptor 1 (brain) | |
| THY1 | 822 | 2 | 2.0 | Thy-1 cell surface antigen | |
| SEMA3A | 781 | 5 | 2.8 | immunoglobulin domain, secreted (semaphorin) 3A | |
| FBN2 | 630 | 2 | 6.4 | fibrillin 2 | |
| ITGA4 | 617 | 3 | 2.7 | integrin (antigen CD49D) alpha 4 subunit of VLA-4 receptor | |
| KCNJ6 | 569 | 0 | 4.2 | potassium inwardly-rectifying channel, subfamily J, member 6 | |
| KIT | 543 | 3 | 7.0 | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | |
| 4 | POSTN | 1541 | 5 | 5.8 | periostin, osteoblast specific factor |
| RUNX2 | 1454 | 5 | 2.9 | runt-related transcription factor 2 | |
| NCAM1 | 1230 | 4 | 3.4 | neural cell adhesion molecule 1 | |
| GATA2 | 1050 | 3 | 3.5 | GATA binding protein 2 | |
| EDIL3 | 901 | 3 | 4.6 | EGF-like repeats and discoidin I-like domains 3 | |
| PAX3 | 840 | 4 | 87.1 | paired box 3 | |
| IRX1 | 768 | 3 | 23.4 | iroquois homeobox 1 | |
| NTNG1 | 760 | 1 | 7.0 | netrin G1 | |
| HAND2 | 752 | 2 | 30.5 | heart and neural crest derivatives expressed 2 | |
| ITGB2 | 663 | 2 | 2.6 | integrin, beta 2 (complement component 3 receptor |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, B.B.; Arianti, R.; Shaw, A.; Vámos, A.; Veréb, Z.; Póliska, S.; Győry, F.; Bacso, Z.; Fésüs, L.; Kristóf, E. FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARγ Specific Regulation: A Transcriptome Analysis. Cells 2020, 9, 987. https://doi.org/10.3390/cells9040987
Tóth BB, Arianti R, Shaw A, Vámos A, Veréb Z, Póliska S, Győry F, Bacso Z, Fésüs L, Kristóf E. FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARγ Specific Regulation: A Transcriptome Analysis. Cells. 2020; 9(4):987. https://doi.org/10.3390/cells9040987
Chicago/Turabian StyleTóth, Beáta B., Rini Arianti, Abhirup Shaw, Attila Vámos, Zoltán Veréb, Szilárd Póliska, Ferenc Győry, Zsolt Bacso, László Fésüs, and Endre Kristóf. 2020. "FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARγ Specific Regulation: A Transcriptome Analysis" Cells 9, no. 4: 987. https://doi.org/10.3390/cells9040987
APA StyleTóth, B. B., Arianti, R., Shaw, A., Vámos, A., Veréb, Z., Póliska, S., Győry, F., Bacso, Z., Fésüs, L., & Kristóf, E. (2020). FTO Intronic SNP Strongly Influences Human Neck Adipocyte Browning Determined by Tissue and PPARγ Specific Regulation: A Transcriptome Analysis. Cells, 9(4), 987. https://doi.org/10.3390/cells9040987